PNAS:一种最新的基于基因测序的染色体组装技术
生物通,2013/01/16
导读:美国伊利诺伊大学的研究人员开发出了一种新方法,能在没有任何已有物理或遗传图谱的情况下,预测出物种染色体的相应组装。这种方法被称为辅助染色体组装,其工作原理就是比较基因组信息和双末端序列信息。相关研究发表在《美国国家科学院院刊》杂志上。
你有没有试过在不知道最终图案的情况下玩拼图游戏?这正是一些基因组研究人员在尝试通过新一代DNA测序数据,拼接成染色体时所面临的同样问题。这些染色体能提供基因组组织和结构变异方面的信息,有助于解析进化历史。为了能拼凑出这些染色体来,科学家们可以通过物理或者遗传图谱完成,但是对于许多物种而言,这种指导性的图谱并不存在。
不过现在,一组来自美国伊利诺伊大学的研究人员开发出了一种新方法,能在没有任何已有物理或遗传图谱的情况下,预测出物种染色体的相应组装。这种方法被称为辅助染色体组装(reference-assisted chromosome assembly,RACA),其工作原理就是比较基因组信息和双末端序列信息。
RACA方法工作原理
“我们设计的这种方法靶向基因组,或者说是基因组保守性,尝试将其进一步融入到进化背景中去,”文章的通讯作者,伊利诺斯大学生物工程系助理教授马健(Jian Ma)解释道,“这样就能分析出其构架,以及密切相关的其它基因组信息。”
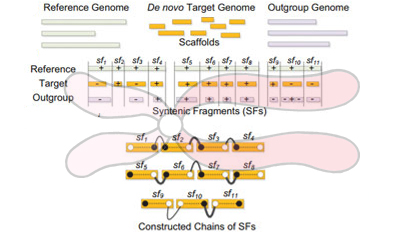 通过BGI研究院科学家们的验证分析,这一研究组预测出了藏羚羊可能的染色体片段组装方法,为了完成这一目标,研究组成员利用BGI的SOAPdenovo组装程序构建出了1434个序列支架,然后重建出了60个羚羊的染色体片段,其中16个片段与牛的染色体片段相似。
通过BGI研究院科学家们的验证分析,这一研究组预测出了藏羚羊可能的染色体片段组装方法,为了完成这一目标,研究组成员利用BGI的SOAPdenovo组装程序构建出了1434个序列支架,然后重建出了60个羚羊的染色体片段,其中16个片段与牛的染色体片段相似。
“在进行程序处理后,基因组质量得到了明显的提高,”马博士解释道,“染色体片段大量减少,连续性延长,并且可以与其他物种进行比较分析了。而且我们之后也能纠正在这一过程中可能出现的组装误差。”
这一研究组面临的主要挑战之一还在于要找到一种能彻底评估分析结果,以及检测其工具的方法,为此研究人员将RACA分析结果,与模拟基因组组装,以及真实基因组组装进行了比较,其中真实基因组组装数据来自约翰霍普金斯大学完成的2012基因组组装金标准评价(GAGE)。
“我们的数据基本上都来自(GAGE)研究,因为这些数据真实,反映了真正的情况,所以可以检测分析工具,”马博士说,“我们分析了他们研究中采用的各种组装结果,结果我们发现,我们可以改善这些结果。”
马博士表示,现在这项技术可以立即被用于类似Genome 10K之类的项目中,这是2009年发起的一项测定万种脊椎动物基因组图谱的项目,其目的在于研究生物多样性和动物进化的机制。
“大多数(基因组研究)都在使用NGS技术,因此我们认为这种方法可以用来系统地改善这些新物种基因组质量”,马博士说。
