杨雪瑞课题组提出使用ribosome profiling数据鉴定基因翻译差异的新型分析方法
2016年4月4日,清华大学生命学院杨雪瑞研究组在Nature Communications杂志发表题为“Genome-wide assessment of differential translations with ribosome profiling data”的研究论文,详细介绍了一种针对全基因组核糖体分析(ribosome profiling)数据的分析方法,Xtail,用于在组学水平对基因的翻译差异进行准确、定量的评估。
mRNA的翻译是基因表达多级调控过程中非常重要的一个步骤。对细胞内RNA与蛋白质合成与降解的定量分析结果显示,从全局的角度来看,翻译这一层级的调控占基因表达调控的50%以上。多项研究表明翻译的异常与细胞生理活动紊乱及多种疾病有密切的联系。长期以来,相比于基因组和转录组的研究,由于实验及分析手段的局限,对mRNA翻译组学的研究一直比较滞后。UCSF Weissman实验室开发的ribosome profiling技术结合了核糖体印迹与深度测序,使翻译在全基因组范围、单个密码子精度的定量成为可能。自发明以来,这一新型翻译组学技术已经被广泛应用于寻找不同实验及生理条件下翻译水平发生显著异常的基因,产生了大量数据。然而,由于数据分析方法的滞后,此前的许多相关研究工作仍然只集中于有限的一些基因,并未充分发挥该高通量分析技术的优势,未能获得较完整准确的基因翻译异常图谱,因此阻碍了我们对相关分子机制在基因翻译层级的系统理解。
杨雪瑞课题组的一个重要研究方向为使用ribosome profiling技术系统分析肿瘤等重要疾病或发育相关的翻译调控机制。在对实验数据的深度分析中,他们发现,由于数据结构的复杂性及多种来源的噪音干扰,现有的针对性数据处理方法存在敏感性差、准确度低的问题。为解决这一问题,充分利用核糖体分析这一前沿技术,杨雪瑞实验室针对ribosome profiling数据构建了新的分析模型,设计了一整套数据处理方法流程, Xtail。Xtail对于翻译异常的基因有非常高的敏感性,并且对数据噪音有高容忍度,保证了极低的假阳性率。在论文中,不管是用于处理模拟数据还是真实数据,Xtail均表现出非常高的准确率,显著超越了现有的其它方法。通过对前列腺癌细胞PC3和人巨噬细胞两套数据的分析,Xtail准确鉴定出具有生物学意义的翻译异常基因集,并在这两种细胞中揭示了一系列新的和肿瘤迁移及巨噬细胞活化相关的翻译异常事件。
总之,Xtail分析流程可以帮助研究人员在全基因组水平更准确、更全面地分析翻译异常调控,为未来的基因翻译相关研究工作提供了高效的系统分析工具。同时,使用Xtail对大量现有ribosome profiling数据的重新分析将提出一系列生物学过程中全面的基因翻译图谱,为理解细胞应对压力刺激、信号传导通路的异常、疾病的发生发展提供更多在基因翻译调控层级的线索。
清华大学生命学院2013级在读博士生肖正涛为本文的第一作者,其他作者为PTN项目博士生邹沁及CLS项目博士生刘昱。清华大学生命学院杨雪瑞研究员为本文的通讯作者。本研究由国家自然科学基金委、青年千人计划、清华-北大生命科学联合中心、清华大学自主科研项目提供经费支持。
原文链接:
http://www.nature.com/ncomms/2016/160404/ncomms11194/full/ncomms11194.html
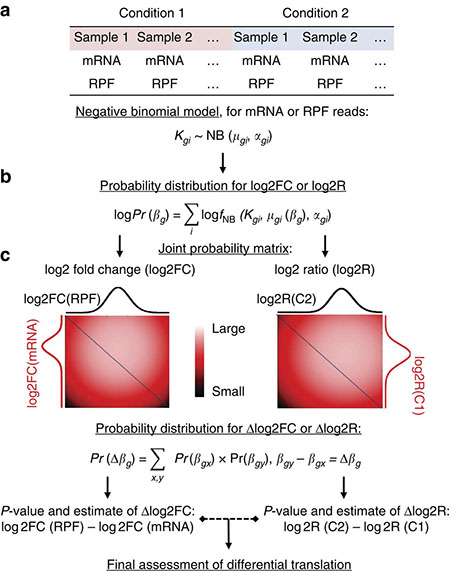
Xtail方法的原理
