一种合成植物激素可抗癌
最近,美国乔治城大学医学中心的研究人员称,有两种药物——模拟一种常见的植物激素,能够有效地导致DNA损伤,并关闭一个主要的DNA修复机制,从而表明它们作为一种抗癌疗法的潜在用途。这项研究结果发表在国际著名杂志《Oncotarget》。
这两种药物——MEB55和ST362,是植物激素独角金内酯的合成版本,独角金内酯是在植物根部产生的,可调节地下根系和地上部枝叶的发育。
乔治城大学的研究人员,首先探讨了这种激素的抗癌特性,自2009年以来他们已经带领了一系列研究表明,独角金内酯的合成版本可抑制乳腺癌、前列腺癌、结肠癌、肺癌和其他多种肿瘤细胞的生长。他们的这项新研究,揭示了这种植物激素的作用机制,以及当它与其他抗癌药物联合使用时对前列腺癌细胞的杀伤作用。
这项研究的高级研究员、乔治城大学护理与健康研究学院人类科学系助理教授、Lombardi 综合癌症中心成员Ronit Yarden博士指出:“MEB55和ST362似乎是非常有前途的药物。我们的研究表明,当与称为PARP抑制剂的抗癌药物共同使用时,这种组合是有效的,并且不伤害正常细胞。”
她的合作者包括以色列农业研究组织(ARO)和意大利都灵大学的研究人员,他们首次合成了MEB55和ST362。使用乔治城大学开发的一项技术——称为条件重新编程的细胞,可使细胞无限增长,研究人员可以在病人的前列腺癌细胞中研究这两种药物。
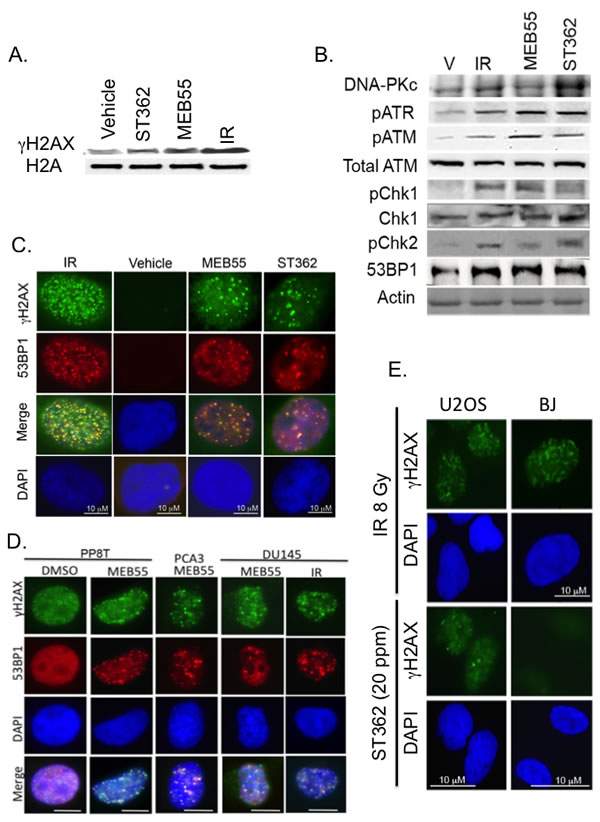
这项研究表明,当MEB55或ST362与PARP抑制剂联合使用时,细胞就会死亡。合成的植物激素会阻止DNA修复过程——发生在细胞的DNA复制之后和细胞分裂之前。PARP抑制剂可关闭另一个修复通路,使癌细胞别无选择,只能死亡。Yarden说:“在癌细胞中,DNA复制错误是特别普遍的,所以,如果没有任何办法修复它们的DNA,这些细胞就会自毁。”
Yarden表示,使用PARP抑制剂的这个想法,来自于其在乳腺癌和卵巢癌中的使用,这些癌症的发展是由于BRCA1或BRCA2基因的突变。当BRCA基因正常时,可控制一种DNA修复途径,但当其突变时,就不能修复基因——合成的植物激素提供了相同的作用。
Yarden表示,她希望很快能够在多种肿瘤动物模型中,对这些药物组合展开试验。
在与癌症的战斗中,开发抗癌药是很多科学家的目标。2016年1月,美国Roswell Park癌症研究所(RPCI)的一个研究团队,发现了一类新的小分子化合物,是为白血病和淋巴瘤开发新的靶向疗法的很好候选对象。这类新的化合物可迫使癌细胞自杀,研究人员将这项研究结果发表在了《Cell Death and Disease》杂志(迫使癌细胞自杀的新抗癌药)。美国勘萨斯大学癌症中心的华人学者发现了一种潜在的抗癌药物,是一种棉籽提取物——棉酚,棉花植物中的一种天然成分,目前正处于前列腺癌临床试验的早期阶段(华人学者用棉籽提取物制备抗癌药)。更有学者对抗癌药进行了巧妙设计,让其发挥更好的疗效,例如,在去年10月份,华人学者顾臻带领的研究团队,首次开发出一种技术,把抗癌药物表面涂覆上来自于患者自身的血小板膜,从而使药物在体内持续的时间更长,可同时攻击原发肿瘤细胞和循环肿瘤细胞,这两种细胞都可以导致癌症的转移。相关研究结果发表在材料科学顶级期刊《Advanced Materials》(顾臻博士:巧伪装抗癌药摧毁肿瘤)。(来源:生物通 王英)
Analogs of the novel phytohormone, strigolactone, trigger apoptosis and synergize with PARP inhibitors by inducing DNA damage and inhibiting DNA repair
Abstract Strigolactones are a novel class of plant hormones produced in roots that regulate shoot and root development. We previously reported that strigolactone analogs (SLs) induce G2/M cell cycle arrest and apoptosis in a variety of human cancer cells and inhibit tumor growth of human breast cancer xenografts in mice. SLs had no significant influences on non-transformed cells. Here we report for the first time that SLs induce DNA damage in the form of DNA double-strand breaks (DSBs) and activate the DNA damage response signaling by inducing phosphorylation of ATM, ATR and DNA-PKcs and co-localization of the DNA damage signaling protein, 53BP1, with γH2AX nuclear foci. We further report that in addition to DSBs induction, SLs simultaneously impair DSBs repair, mostly homology-directed repair (HDR) and to a lesser extent non-homologous end joining (NHEJ). In response to SLs, RAD51, the homologous DSB repair protein, is ubiquitinated and targeted for proteasomal degradation and it fails to co-localize with γH2AX foci. Interestingly, SLs synergize with DNA damaging agents-based therapeutics. The combination of PARP inhibitors and SLs showed an especially potent synergy, but only in BRCA1-proficient cells. No synergy was observed between SLs and PARP inhibitors in BRCA1-deficient cells, supporting a role for SLs in HDR impairment. Together, our data suggest that SLs increase genome instability and cell death by a unique mechanism of inducing DNA damage and inhibiting DNA repair.
原文链接:http://www.impactjournals.com/oncotarget/index.php?journal=oncotarget&page=article&op=view&path%5B%5D=7414&path%5B%5D=21308
