PNAS:华东理工大学王启要教授揭示细菌产生抗生素抗性的新机制
2016年1月4日,国际著名学术期刊《美国国家科学院院刊》(Proceedings of the National Academy of sciences of the United States of America)在线发表华东理工大学生物反应器工程国家重点实验室和美国康涅狄格大学李露(Lu Li)教授合作发现的有关细菌感应β-内酰胺类抗生素并产生抗性的新机制。华东理工大学生物工程学院阿华所青年教师王启要教授为此论文共同第一作者。
导致人类和海洋动物致病的海洋病原性弧菌(非O1-O139霍乱弧菌、副溶血弧菌、溶藻弧菌和鳗弧菌等)天然具有β-内酰胺类抗生素的抗性,然而其产生抗性的信号传导和调控机制不清楚。经典情况下,β-内酰胺类抗生素干扰革兰氏阴性细菌细胞壁合成所需转肽酶(也称为青霉素结合蛋白)的活性。 相应地,细菌由于细胞壁受损,胞壁肽在周质空间中积累并转运至细胞内,结合至转录因子AmpR后活化后者的转录活性并激活 β-内酰胺类抗生素抗性基因(β-内酰胺酶基因)的表达。β-内酰胺酶降解内β-酰胺环并使抗生素失活,因而细菌产生对该类抗生素的抗性。
研究中首次发现双组分系统BrsK-BrrR能以不依赖经典细胞壁损伤应激的机制感应胞外β-内酰胺类抗生素并直接调控抗性基因表达(图A)。其中组氨酸激酶BrsK胞外的口袋结构(图B红色部分)能直接结合β-内酰胺类抗生素,并通过BrsK胞内组氨酸位点与BrrR的天冬氨酸位点的磷酸化-去磷酸化机制,将信号传递至胞内调控抗性基因表达。相比于经典信号转导途径,该机制依赖双组分系统直接识别并结合抗生素,响应迅速并不需要细胞壁损伤信号,在细菌抗生素抗性机制进化中具有明显优势。该发现为将来针对病原性弧菌新型抗菌药物的理性设计和β-内酰胺类抗生素的改进奠定了基础。
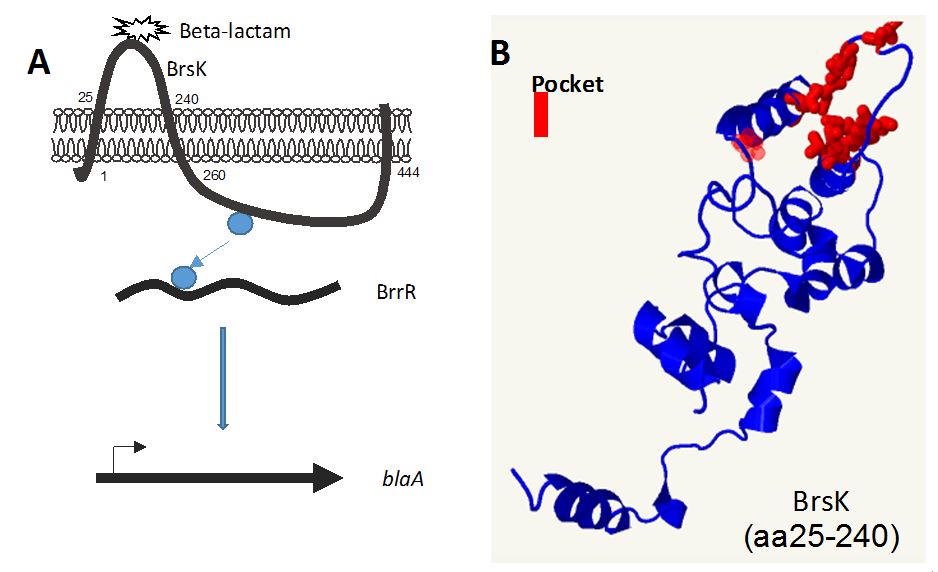
图片:BrsK-BrrR直接调控抗性基因表达
原文链接:
Sensor histidine kinase is a β-lactam receptor and induces resistance to β-lactam antibiotics
原文摘要:
β-Lactams disrupt bacterial cell wall synthesis, and these agents are the most widely used antibiotics. One of the principle mechanisms by which bacteria resist the action of β-lactams is by producing β-lactamases, enzymes that degrade β-lactams. In Gram-negative bacteria, production of β-lactamases is often induced in response to the antibiotic-associated damage to the cell wall. Here, we have identified a previously unidentified mechanism that governs β-lactamase production. In the Gram-negative enteric pathogen Vibrio parahaemolyticus, we found a histidine kinase/response regulator pair (VbrK/VbrR) that controls expression of a β-lactamase. Mutants lacking either VbrK or VbrR do not produce the β-lactamase and are no longer resistant to β-lactam antibiotics. Notably, VbrK autophosphorylation is activated by β-lactam antibiotics, but not by other lactams. However, single amino acid substitutions in the putative periplasmic binding pocket of VbrK leads its phosphorylation in response to both β-lactam and other lactams, suggesting that this kinase is a β-lactam receptor that can directly detect β-lactam antibiotics instead of detecting the damage to cell wall resulting from β-lactams. In strong support of this idea, we found that purified periplasmic sensor domain of VbrK binds penicillin, and that such binding is critical for VbrK autophosphorylation and β-lactamase production. Direct recognition of β-lactam antibiotics by a histidine kinase receptor may represent an evolutionarily favorable mechanism to defend against β-lactam antibiotics.
作者:王启要
