3.4 miRNA与lncRNA的生物信息学预测
1 miRNA生物信息学预测
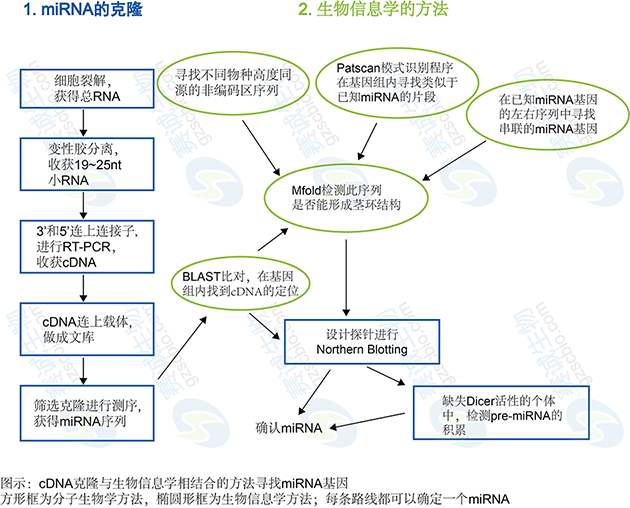
图1 生物信息学在miRNA研究中的应用
当开始研究一基因是否为一个miRNA调控的靶基因时,可以用不同的生物信息学计算方法来分析每个序列(如mRNA的3'-UTR区序列),这些计算方法采用不同的参数来预测一个给定的靶mRNA内具功能性miRNA结合位点的可能性。由于每种计算方法的有效性不同,下面3种计算方法应该被用来预测miRNA结合位点:miRanda、TargetScan和PicTar.这3种计算方法都允许研究者输入一个基因符号,这些计算方法将计算此基因内所有预测的miRNA结合位点。此外,这些计算方法可测定一个给定的miRNA所有的靶mRNA.因为不同的计算方法会预测出不同的miRNA结合位点,所以同时使用多种计算方法进行预测非常必要。值得注意的是,尽管miRNA结合位点在不同物种间的保守性是各种不同计算方法的组成部分,但并不是一个功能性位点所必需的。由于不同计算方法预测的结果存在很大的差异,如何确定哪些预测的结合位点需要进一步的实验验证成为研究者要面临的一个难题。作者认为至少这3种计算方法中的2种计算方法均预测到的miRNA结合位点,有必要进一步用实验验证。
因为很多经种子序列匹配预测的miRNA靶经体内验证实验证实并不是真的miRNA靶,为了起始一步减少预测到的抑制一给定的靶mRNA表达的miRNA的数量,进一步的程序分析是有必要的。结构特征控制着miRNA/mRNA间的相互作用的观点已被越来越多的人所接受。例如,一个RNA分子的大部分结构是高度复杂性的,只有特定的单链区域允许miRNAs接近并与互补位点结合。因此,复杂的RNA二级结构可能阻止miRNA/mRNA的相互作用。最近有研究证实,绝大部分已证实的靶的一个共同特征是优先与基于热动力学在RNA分子中容易接近且没有复杂二级结构的3’-UTR区中的位点。由于RNA可接近性可能是靶识别的一个关键特征,所以有必要采用mFold软件测定预测到的miRNA结合位点5’端和3’端各70个核苷酸的自由能,当其低于平均随机自由能时提示此位点允许miRNA接近并结合[20].这些允许miRNA接近并结合起来的位点,有必要进一步用实验进行验证。
在不同物种中成熟miRNA均是从具有茎环状二级结构的前体加工而来,具有较大的序列同源性。克隆到的miRNA序列通过检索基因组数据库找到在基因组中的位置,在和周围基因组序列比较中发现他们同样具有相似的前体结构,多位于编码基因间或内含子反向重复区域。一些miRNA基因在进化上具有高度保守性,此为生物信息学筛选的基础。该方法根据比较基因组学原理,并结合生物信息软件在已测序基因组中进行搜索比对,根据同源性的高低再进行RNA二级结构预测,将符合条件的候选miRNA与已经通过试验鉴定的miRNA分子进行比较分析,最终确定该物种miRNA的分步及数量。目前国际上较为普遍使用的两个计算机分析工具是miRseeker和miRscan,前者已用于果蝇及昆虫基因组候选基因的系统分析,后者则用于线虫和脊椎动物候选基因的分析。这两个工具已经成功鉴定出了大量的miRNA基因并通过了实验证实。由于miRseeker和miRscan的高灵敏度,它们已用于人类miRNA基因的寻找。由于该方法只能用于已完成基因组测序的物种,而那些未完成测序的物种就无能为力,而且由于miRNA前体长度的可变性,故用计算机方法寻找新基因具有一定的遗漏性,所以目前大多数实验室将计算机分析与实验方法结合使用,使得miRNA的发现量成几何级数增长。目前日益发展的微阵列技术也在筛选miRNA基因方面显示了极大的潜力。
随着疾病特异性的miRNAs不断被鉴定,对感兴趣的疾病通路中的新靶基因进行验证可能催生新的治疗策略。因此,能够鉴定和验证miRNA/mRNA靶配对具有极其重要的意义。尽管生物信息学方法和自由能分析并不完美,但可使作者能够对推测的miRNA/mRNA靶配对进行鉴定。一旦生物信息学方法预测成功,可以通过以下4条标准验证miRNA/mRNA靶配对的真实性。(1)miRNA/mRNA靶相互作用得到验证。(2)miRNA/mRNA共表达。(3)给定miRNA对其蛋白表达有可预测的影响。即用此miRNA的类似物可减少靶基因表达水平,而用此miRNA特异性抑制剂可增加靶基因的表达水平。(4)miRNA介导靶基因表达的调控导致相应的生物学功能的改变。
2 LncRNA的生物信息学预测
对lncRNA进行鉴定时,采取的策略是收集不同类型的数据(包括polyA RNA sequencing、nonpolyA RNA sequencing、表观遗传信号值、编码可能性、保守性和RNA结构等),并对其进行分析。例如CDS的RNA-seqpolyA的表达值比较高,而ncRNA的RNA-seqnon-polyA表达值比较高。通过对不同类型数据的整合,还可以进一步得到不同类型基因元素的网络调控关系。
对lncRNA进行综合分析的一般流程如下:(1)将基因组划分成小的单位(bin),根据Gencode的注释信息对每个bin进行注释;(2)分别计算每个bin的特征值,这些特征值包括序列保守性、结构稳定性、RNA表达值、组蛋白修饰、转录因子结合等;(3)利用机器学习的模型,将lncRNA与其他基因类别区分开,并且对新的lncRNA进行预测。

图2 利用数据整合对lncRNA进行鉴定

图3 lncRNA综合分析方法流程示例
有的时候我们的专业知识不足以完成分析和预测。尤其在面对高通量数据时,从中挖掘有用的信息尤为关键。这时可以用到机器学习(machinelearning)的方法,令机器自动分析数据,比如特征提取或是分类。机器学习应用在生物信息学主要有两大分支,即监督学习(supervisedlearning)和非监督学习(unsupervisedlearning)。在监督学习问题中,每个数据拥有一个对应标签,我们希望通过数据建立一个模型,根据数据预测标签。传统的监督学习方法包括线性判别分析(LDA)、决策树(decisiontree)、最近邻法(nearestneighbor)和神经网络(neuralnetwork)。20世纪90年代后,诞生了一批很有影响力的工作,包括支持向量机(SVM)、Adaboosting和随机森林(randomforest),相比于传统的方法,上述方法更好地处理了过拟合(overfitting)的问题,从而在实际应用中有很好的预测效果。
LncRNA研究是基因组时代重要的科学前沿,因为它有可能揭示一个全新的由RNA介导的遗传信息表达调控网络,从不同于蛋白质编码基因的角度来注释和阐明基因组的结构与功能,并为人类的疾病研究和治疗提供新的思路和方法。同时,新一代测序技术的发展也为鉴定lncRNA的计算机方法提供了强大的支持。以下是整理的长非编码RNA(lncRNA,lincRNA)数据库资源列表(按字母排序)。国内外长非编码RNA的研究刚刚兴起,希望这资源对国内的非编码RNA的研究者有所帮助。
(1) ChIPBase:提供长链非编码RNA的表达图谱和转录调控的全面鉴定和注释。整合了高通量的RNA-seq鉴定的lncRNA及其表达图谱和ChIP-Seq实验技术鉴定的转录因子结合位点。
网站:http://deepbase.sysu.edu.cn/chipbase/
更新:2012年11月
(2)LNCipedia:对人类的长链非编码RNA的序列和结构全面的注释。
更新:2012年7月
(3)lncRNAdb:提供有生物学功能的长链非编码RNA的全面注释。这是长链非编码RNA研究领域的大牛John mattick实验室构建的网站。
更新:2011年7月
(4)LncRNADisease:提供了文献报道的疾病相关的长链非编码RNA的注释。
网站:http://cmbi.bjmu.edu.cn/lncrnadisease
更新:2012年7月
(5)NONCODE:提供对长链非编码RNA的全面注释,包括表达和该团队开发的ncFANs计算机软件预测的lncRNA功能。这是非编码RNA研究的知名数据库,已经更新到第三版。
更新:2012年1月
(6)NRED: 提供人和小鼠的长链非编码RNA在芯片数据的表达信息。这也是John mattick实验室构建的网站。
网站:http://jsm-research.imb.uq.edu.au/nred/
更新: 2009年
作者:广州赛诚生物
