仅根据DNA序列即可确定表观基因组情况

一种新的计算方法可以根据DNA序列来预测表观遗传学标志和DNA甲基化模式。
人们长久以来都拥有一个遥不可及的目标——根据某基因位点上的DNA序列,就能够预测出该位点上的基因调控活性。在本期的《自然方法》(Nature Methods)期刊中,Whitaker等人朝着这一目标迈出了一大步。他们构建出了一系列计算模型,只需要利用DNA序列数据,就可以非常准确地预测出组蛋白标志和DNA甲基化的位置。这些前所未有的研究结果可以证明:细胞的大部分表观遗传状态都来源于自身的基因组序列。Whitaker等人在一般细胞和特殊细胞中,确定了一些特殊的、可能会对特定表观遗传学标志的存在与否产生巨大影响的DNA序列模体。
同一有机体内的细胞通常都具有完全相同的基因组序列,但是不同类型的细胞却呈现出完全不同的基因表达情况。每个细胞会利用表观遗传因子,在特定的基因位点上做上转录激活和抑制的标记。这些表观遗传因子包括DNA自身的甲基化以及组蛋白(即可将DNA组装成染色质的蛋白质)的共价修饰,其中被研究得最多的组蛋白修饰是核心组蛋白H3赖氨酸残基的甲基化和乙酰化。这些组蛋白修饰具有以下功能:传递细胞身份标识、激活和抑制转录过程、对重要的基因调控元件(例如转 录起始位点和增强子)进行标记。在此我们将所有组蛋白修饰和DNA甲基化的分布情况作为细胞的表观遗传状态。
同一类型的细胞在发育过程中会倾向于出现相类似的表观遗传状态。为了能够在任意基因位点上重新生成细胞类型特异性的表观遗传状态,细胞需要用到两种信息来源。这两种信息来源包括:该基因位点上曾经出现过、昙花一现的表观遗传学特征,以及更为稳定的DNA序列。由于细胞在发育过程中仅仅凭借这些信息就能够很可靠地重新生成自身的表观基因组,因此我们应该也能够采用计算模型,根据相同的信息来预测细胞的大部分表观遗传状态。直到如今,人们才真正地认识到了这种理论上的可能性。
Whitaker等人研发出了一套名为Epigram的软件应用程序,可以根据DNA序列模体的信息来预测DNA甲基化和组蛋白修饰的情况。他们利用DNA序列数据、染色体免疫共沉淀测序(chromatin immunoprecipitation–sequencing, ChIP-seq)数据和methylC-seq数据构建了这一计算模型。在特定类型的细胞中,DNA序列模体与细胞特异性表观遗传标志之间存在着关联,而他们根据这种关联来鉴定这些DNA序列模体。他们也表明,有一些DNA序列模体往往会专门出现在表观遗传标志富集区域的中心或边缘。
截止到投稿为止,这项研究工作首次直接证明了表观遗传学修饰具有较高的可预测性:我们仅凭借DNA序列就可以准确预测表观遗传学修饰情况。以往的研究工作指出,DNA序列模体断裂与组蛋白H3第27位赖氨酸乙酰化(H3 lysine 27 acetylation, H3K27ac)水平的变化之间存在关联,但是Epigram却能够用更少的模体来解释更多的H3K27ac变异。Benveniste等人在最近发表的另一篇论文中也利用序列信息来预测表观遗传状态。尽管Epigram能够从DNA序列中提取出更有预测性的信息,但是Benveniste等人的方法却稍微超越了Epigram的性能:他们利用ChIP-seq数据,可以对存在有转录因子的区域进行预测。Epigram已经发现了许多具有预测价值的序列模体,但是由于这些模体与那些已经公开发表的转录因子结合模体非常相似,因此这两项研究共同证明:已知的转录因子在促进细胞形成表观遗传状态上发挥了重要的作用。尽管如此,但是如果我们能够在不依赖任何基因位点特异性生化数据(例如ChIP-seq数据)的情况下来预测细胞表观遗传状态的话,这将更加令人印象深刻。这种预测方法的应用范围会非常广泛,甚至在没有额外的生化数据的情况下可以应用。
Epigram之所以能够区别于以往的预测方法,主要是因为研究者已经细致彻底地消除了一些混杂因素的干扰。Whitaker等人利用随机森林分类算法(Random Forest classifier),找出了某种表观遗传标志富集区域附近的序列(前景)与其它序列(背景)之间的差异。Epigram采用一种DNA序列集平衡方法,确保前景序列和背景序列都拥有相近的长度和G+C含量。如此一来,研究者们就不会主要根据这些简单的特征来进行分类,而是根据序列模体来进行分类了。Epigram预测法也设置了一个独立的步骤,在测序过程中校正G+C含量所产生的偏倚。
尽管Whitaker等人确定了与表观遗传标志有关的DNA序列模体,但是他们并未证明这些模体是否会引起表观遗传特征的蓄积。我们可以利用基因组编辑技术或其他实验技术,来证明他们所确定的DNA序列模体在“谱写”表观遗传标志上是否发挥了关键作用(图1)。Whitaker等人的论文为此类研究以及其它表观遗传状态促进因素的机制研究开辟了新的道路。
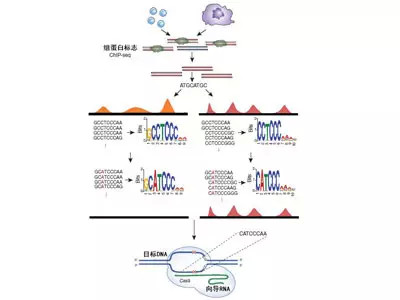
图
1 细胞类型特异性的表观基因组编辑实验。Epigram利用两类细胞的H3K27ac
ChIP-seq数据,就能够为每种细胞的H3K27ac修饰创建序列模型。我们利用Epigram和这些专门创建的模型,就能够在其中一类细胞中寻找到导致H3K27ac富集点敲除的序列改变。如果我们首先应用基因组编辑方法,然后采用ChIP定量PCR技术,就能够证实“表观遗传敲除(epiknockout)”能够像预期的那样发挥作用。
本文来源于:生物360
欢迎关注中科紫鑫人事招聘相关信息:http://www.ngscn.com/index.php/Job/employ
