Genome Res:全基因组测序追踪耐药细菌传播机制
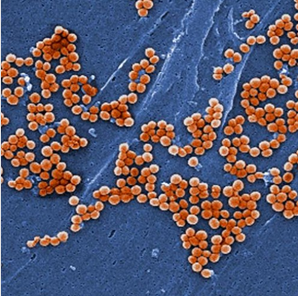
耐甲氧西林金黄色葡萄球菌(MRSA)是一种常见的引发院内感染的致病菌,其也是资源不足医院感染的一大负担,此前当资源较好的临床机构运用全基因组测序来追踪MRSA的扩散时,针对有限的感染控制的传播动力学常常并不清楚,近日,来自剑桥大学的研究人员就利用全基因组测序的技术揭示了高传播率的资源受限医院中MRSA的扩散,相关研究发表于国际杂志genome Research上。
研究者对来自泰国东北部的一家医院的两个加强医疗病房(ICUs)的病人进行了3个多月的MRSA的监测,在监测期间46名病人和5名工作人员至少一次检测出MRSA阳性;而不同MRSA菌株之间的遗传相似性也阻碍了传统低分辨率基因分型技术来对人群之间的传播进行区分,因此全基因组测序技术就派上了用场,研究者利用其对76个分离自病人机体的菌株进行研究。
研究者Sharon Peacock说道,基于金黄色葡萄球菌的全基因组测序的系统发育树的一大显著特征就是多个不同进化枝的出现,这就表明,相同菌株谱系的多个分枝或许在同一时间里在医院进行循环。基于对MRSA分离株的基因组改变对单一菌株进行研究将帮助研究人员推理出感染病人之间的MRAS传播路径,研究者的长期目标就是利用对MRSA的全基因组分析来制定感染的控制策略,而当前文章中阐明的MRSA传播程度指示研究人员应该优先改善个体的手部卫生。
全基因组测序同时也揭示在超过3个月的研究时间里ICU中MRSA的分化枝是动态变化的,随着研究人员深度测序的开展,他们也发现,尽管所有的菌株都具有相同的分化枝,但是其之间还是存在一些微小的遗传差异,这就揭示了在单一的载体中细菌仍具有多样性;当前的研究结果表明,揭示MRSA的传播网络仍然需要测定宿主机体中细菌的多样性。
原文摘要:Methicillin-resistant Staphylococcus aureus (MRSA) is a major cause of nosocomial infection. Whole-genome sequencing of MRSA has been used to define phylogeny and transmission in well-resourced healthcare settings, yet the greatest burden of nosocomial infection occurs in resource-restricted settings where barriers to transmission are lower. Here, we study the flux and genetic diversity of MRSA on ward and individual patient levels in a hospital where transmission was common. We repeatedly screened all patients on two intensive care units for MRSA carriage over a 3-mo period. All MRSA belonged to multilocus sequence type 239 (ST 239). We defined the population structure and charted the spread of MRSA by sequencing 79 isolates from 46 patients and five members of staff, including the first MRSA-positive screen isolates and up to two repeat isolates where available. Phylogenetic analysis identified a flux of distinct ST 239 clades over time in each intensive care unit. In total, five main clades were identified, which varied in the carriage of plasmids encoding antiseptic and antimicrobial resistance determinants. Sequence data confirmed intra- and interwards transmission events and identified individual patients who were colonized by more than one clade. One patient on each unit was the source of numerous transmission events, and deep sampling of one of these cases demonstrated colonization with a “cloud” of related MRSA variants. The application of whole-genome sequencing and analysis provides novel insights into the transmission of MRSA in under-resourced healthcare settings and has relevance to wider global health.
作者:网络
