中国学者获碳-氢键官能团化新突破 助力药物研发
杂环化合物广泛存在于药物分子中,在药物合成和发现过程中扮演着举足轻重的作用,这是因为杂环的存在不仅能够影响药物分子与受体之间的相互作用,而且有利于提高药物分子的溶解度。因此,如何快速构建杂环分子骨架并高效地进行结构多样性合成,受到化学家和药物工业界的极大关注。如果通过一步简单的碳-氢键活化对杂环化合物进行精准的官能团化,可大大缩短药物分子的合成步骤,实现结构多样性分子的快速合成与修饰,使得快速构建庞大的药物分子库成为可能,将对药物的筛选和发现起到巨大推动作用。
然而杂环化合物中存在的氮、硫、磷等杂原子与过渡金属催化剂能够产生非常强的配位作用,这种强配位一方面可能会导致催化剂的失活,另一方面由于配位原子的导向作用,使得碳-氢键活化只能发生在其邻位,从而使得在其它位置的碳-氢键高选择性活化转化极具挑战性,也大大限制了碳-氢键活化方法在药物分子的多样性合成及结构修饰中的应用(图一)。因此,如何克服杂环中的配位杂原子对碳-氢键选择性活化的不利影响是该领域亟待解决的一大难题。
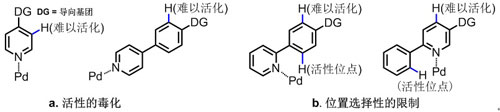
图一 杂环对导向C–H官能团化的影响
中国科学院上海有机化学研究所金属有机化学国家重点实验室余金权教授、戴辉雄博士成功地解决了上述问题。他们以N-甲氧基甲酰胺为导向基团,采用零价钯作为催化剂,通过最为绿色环保的空气为氧化剂,现场生成具有催化活性的二价钯物种(图二),巧妙地避免了杂环中强配位原子对碳-氢键活化的导向作用(图三),抑制了杂环邻位的碳-氢官能团化,使得碳-氢键活化和官能团化能够高选择性地发生在其它位置,实现了杂环化合物碳-氢键官能团化新突破,打破了碳-氢键活化中传统的选择性规律,有望在药物分子多样性合成及修饰方面的实现应用(图四)。该工作在最近一期《自然》(Nature)杂志上以 “突破杂环导向碳-氢官能团化的局限性”(Overcoming the Limitations of Directed C-H Functionalizations of Heterocycles)为题在线发表(Nature 2014, DOI: 10.1038/nature13885)。据检索,这是我国大陆地区有机合成化学领域科学家在《自然》杂志上发表的第一篇论文。
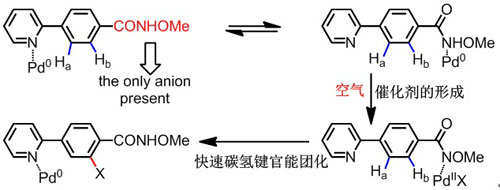
图二 方法:阴离子导向基团促进的现场生成二价钯催化剂
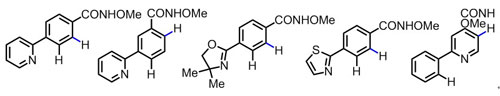
图三 克服强配位的杂环对位置选择性的影响
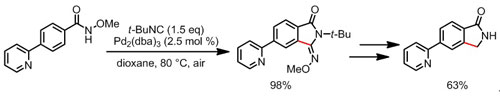
图四 含杂环官能团的内酰胺化合物的形成
据戴辉雄介绍, 该催化体系实现了56个杂环化合物的碳-氢官能团化反应,显示了对杂环中N、S、P等原子的容忍性和底物结构的兼容性,很好地克服了这些杂原子对反应区域选择性的影响。该反应表现出很高的催化效率以及原子经济性,特别是采用空气作为氧化剂,以0.5 mol% Pd2(dba)2作为催化剂在半小时内就能够完成,具有很好的实用价值。因此,这一新方法的实现对于有机合成化学和药物化学的发展将可能起到较大的推动作用。
上述研究得到了中组部千人计划创新人才短期项目、国家自然科学基金委和中国科学院的大力资助。(来源:生物360)
Overcoming the limitations of directed C–H functionalizations of heterocycles
Abstract In directed C–H activation reactions, any nitrogen or sulphur atoms present in heterocyclic substrates will coordinate strongly with metal catalysts. This coordination, which can lead to catalyst poisoning or C–H functionalization at an undesired position, limits the application of C–H activation reactions in heterocycle-based drug discovery, in which regard they have attracted much interest from pharmaceutical companies. Here we report a robust and synthetically useful method that overcomes the complications associated with performing C–H functionalization reactions on heterocycles. Our approach employs a simple N-methoxy amide group, which serves as both a directing group and an anionic ligand that promotes the in situ generation of the reactive PdX2 (X = ArCONOMe) species from a Pd(0) source using air as the sole oxidant. In this way, the PdX2 species is localized near the target C–H bond, avoiding interference from any nitrogen or sulphur atoms present in the heterocyclic substrates. This reaction overrides the conventional positional selectivity patterns observed with substrates containing strongly coordinating heteroatoms, including nitrogen, sulphur and phosphorus. Thus, this operationally simple aerobic reaction demonstrates that it is possible to bypass a fundamental limitation that has long plagued applications of directed C–H activation in medicinal chemistry.
原文链接:http://www.nature.com/nature/journal/vaop/ncurrent/pdf/nature13885.pdf
