计算生物学所提出高效新方法定位具进化优势的基因突变
8月18日,国际著名学术期刊Molecular Biology and Evolution在线发表了中国科学院上海生命科学研究院计算生物学研究所博士生汪敏先的最新研究论文“Detecting Recent Positive Selection with High Accuracy and Reliability by Conditional Coalescent Tree”。该论文介绍了一种新的群体遗传学数据分析方法。该方法使用基因组测序数据,可以高效可靠地识别近期受到正选择的遗传多态性位点。新方法的有效性得到了计算机模拟和实际数据研究的双重验证。
群体遗传学和考古学研究表明现代人类的直接祖先来自非洲。自约十万年前开始,他们由非洲大陆逐渐迁徙扩散到欧洲、亚洲和美洲等世界各地。在这一迁徙过程中,人类祖先跨越不同的自然地理区域,经历了多样的气候、多变的食物来源、以及不同传染性疾病等自然力量的挑战。人群中具有某些特定基因变异的个体比其他个体能够更好地应对这些挑战,从而留下更多的后代,导致这些特定的基因变异类型在人群中的频率显著上升。遗传学上把这种现象称为“正选择”。识别这些近期受到“正选择”的基因变异,并进一步研究其背后生物学意义,对从生物学角度了解人类自身具有重大意义。然而,从基因组数以百万计的遗传变异位点中准确找出这些受正选择影响的关键变异位点并非易事。
在何云刚和金力的指导下,汪敏先基于条件溯祖理论成功建立了新的计算方法,在对受正选择的关键遗传变异的识别和准确定位上有所突破。新方法的定位准确率较之前的方法提高了约20~40%,统计效力及稳健性也获得明显改善。实例研究中,新的计算方法准确识别了ADH1B、MCM6、APOL1和HBB基因中已得到生物学实验证实的关键基因突变(见图)。这样精确的定位能力为深入开展“正选择”有关的生物学功能研究带来了极大便利。新方法的计算速度远远领先于最流行的正选择检查方法iHS,因而非常适合用于大规模高通量测序数据分析。该研究工作得到了来自国家自然科学基金委和中国科学院的基金支持。(计算生物学所)
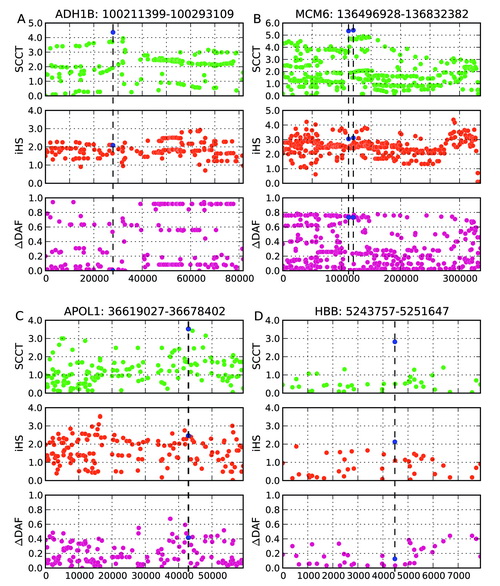
新方法(SCCT)准确定位关键基因突变
