我室康振生院士团队在小麦抗条锈病基因发掘方面取得重要进展
2020年7月17日《Plant Biotechnology Journal》杂志在线发表了我室康振生院士植物免疫团队的题为“A large-scale genomic association analysis identifies the candidate causal genes conferring stripe rust resistance under multiple field environments”的研究成果。该团队的吴建辉博士、蔚睿硕士、王海英硕士为该论文的共同第一作者,康振生教授、韩德俊教授、曾庆东副教授为该论文的共同通讯作者。该项研究得到国家自然科学基金、博士后基金和开放课题等基金的资助。
小麦条锈病是全球范围内广泛流行的活体寄生性真菌病害,一直严重威胁我国小麦安全生产。培育、推广抗病小麦品种一直是最为经济有效且绿色环保的防控措施。然而条锈菌可通过突变、异核作用以及有性生殖等诸多变异途径产生大量新毒性小种,从而导致小麦抗病品种投入生产后3~5内“丧失”抗性。大量研究表明,成株期抗病性往往具有持久性特点,且从已克隆的抗病基因分析发现这类基因序列相对保守,在中国春参考基因组上能找到同源序列。因此,可以通过分析中国春基因组上后选区间的基因来初步鉴定成株期抗条锈病候选基因。
该团队从世界范围内收集的5000多份小麦种质中,通过分子标记遗传多样性挑选出1500份代表种质,进行小麦660K芯片基因分型。另外,利用60多份普通小麦的重测序数据辅助加密分型信息。这为通过全基因组水平进行抗锈病基因位点的发掘、鉴定奠定了材料和数据基础。利用前人开发的全基因关联分析(GWAS)模型,基于对每个核苷酸多态性的功能重要性进行评估,快速鉴定候选基因(Yano et al., 2016)。基于以上研究基础和思路,本研究对411份春小麦种质进行多年多点条锈病抗性鉴定,结合660K芯片基因型数据,通过全基因组关联分析快速发掘292个与抗病表型显著关联的SNP位点,分布于19个区域中,其中14个区域为已知的基因/QTL且被广泛应用于育种中。

图一 全基因组关联分析定位抗条锈位点(曼哈顿图)
随后我们在已克隆的基因Yr18内部及上下游发现存在这样的SNP变异,说明在小麦中,针对染色体某些特定区域,利用高密度SNP芯片进行候选区间的关联分析是可行的。
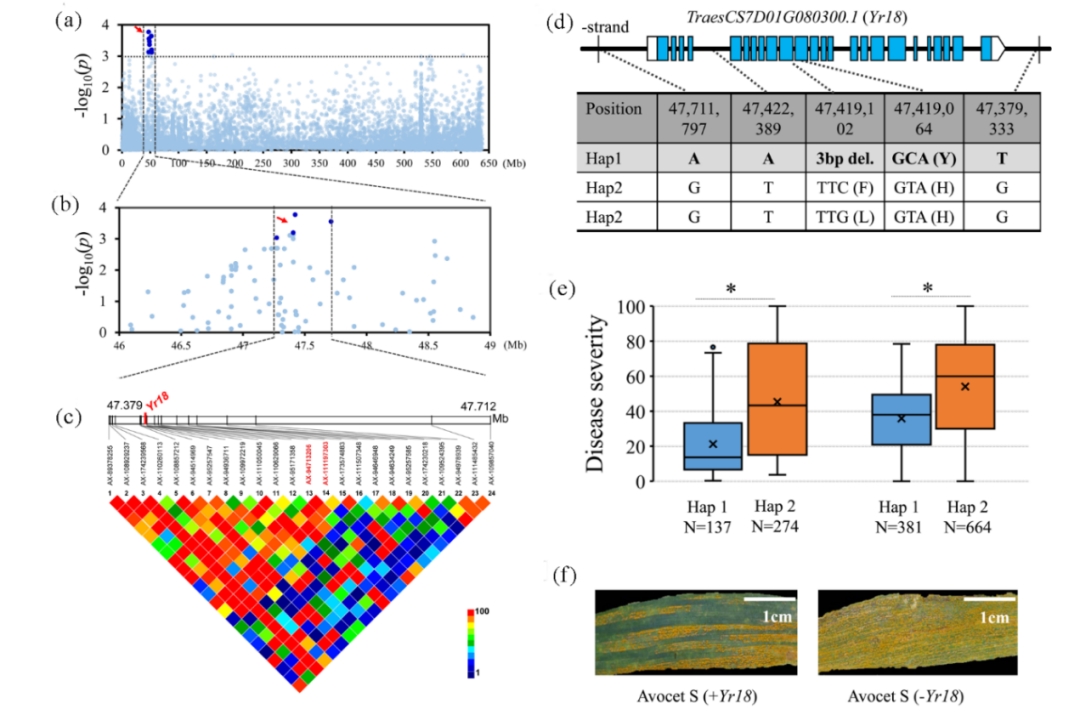
图二 利用关联分析对已克隆的Yr18基因进行验证
利用这个分析思路,很快对2BL上的主效位点YrSnb.1-2BL进行了分析,并通过另外一个自然群体(个数超过1000)的关联分析,有效的缩小了候选区间。结合芯片及重测序数据,总共筛选到约800个多态性SNP变异,涵盖了该区间的12个高可信基因。分析发现大部分显著性的SNP变异都集中在TraesCS2B01G512900, TraesCS2B01G513000和 TraesCS2B01G513100三个基因周围,然而只在TraesCS2B01G513100的编码区发现了3个显著性序列变异(−log10P≥ 4.85),这3个SNP的变异引起了氨基酸的改变。另外,还有4个显著性的SNP变异,其中2个位于TraesCS2B01G513000的启动子区域,另外2个分别位于TraesCS2B01G512900和TraesCS2B01G513100的下游。有趣的是,这3个基因都编码丝氨酸/苏氨酸激酶(serine/threonine protein kinases, STPKs),以往研究表明,STPK确实参与小麦对真菌病害的抗性反应。为了进一步确认候选基因,对候选区间的12个基因都进行成株期小麦-条锈互作表达分析,发现只有TraesCS2B01G513100在抗感对照中差异表达。另外,利用四倍体小麦Kronos的EMS诱变的突变体进行条锈菌接种验证,发现TraesCS2B01G513100中的两个突变体感病性要高于野生型,其他的无显著差异。以上结果表明TraesCS2B01G513100很有可能就是参与条锈抗病反应的重要候选基因成员。此外,对该候选区间进行了单倍型分析,发现含有“TGCGGT”单倍型的材料具有良好的抗病性,为此开发了相应的育种分子辅助选择标记。
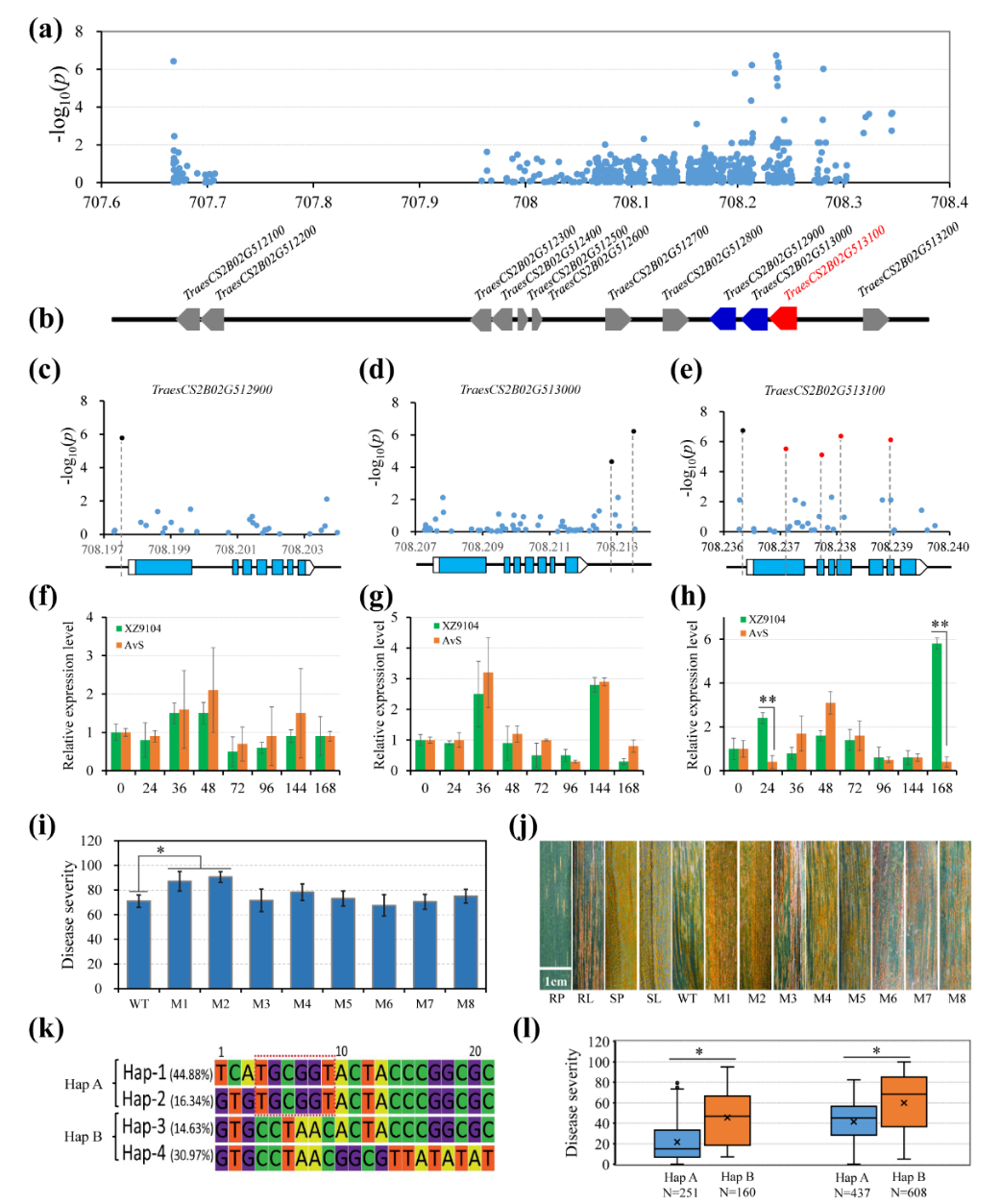
图三 YrSnb.1-2BL候选基因分析、初步验证和单倍型分析
