我校在改进细菌16S rRNA基因高通量测序方法研究上有新进展
南湖新闻网讯(审核人 宋露洋)近日,我校作物遗传改良国家重点实验室谢卡斌课题组在Microbiome发表了题为“Engineering CRISPR/Cas9 to mitigate abundant host contamination for 16S rRNA gene-based amplicon sequencing”的研究论文,论文报道了一种改进的细菌16S rRNA基因高通量测序方法。
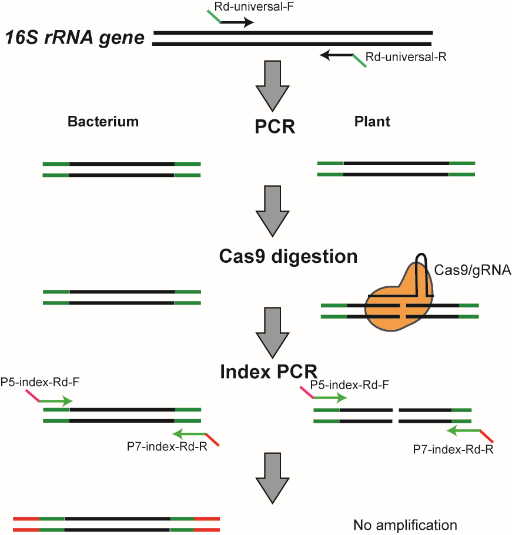
Cas-16S-seq方法示意图
16S-seq是微生物组学研究最常用的方法,而大量叶绿体和线粒体序列污染是16S-seq分析植物微生物组的一大难题。该研究巧妙地利用CRISPR/Cas9系统靶向消除16S-seq测序文库中的宿主序列,保留完整的细菌16S rRNA基因信息,提高了16S-seq分析植物样品的灵敏度和效率。
微生物组对动植物健康具有重要意义。对植物微生物组的深入研究将为调控植物生长、增强作物抗逆性、减少植物病害以及促进农业可持续发展提供新的策略。16S-seq是研究细菌群落结构的常用方法。然而,线粒体和叶绿体存在和细菌同源的16S rRNA基因,因此植物样品的16S-seq数据中会有最高可达99%以上的宿主16S rRNA序列,严重影响了16S-seq在植物中的应用。
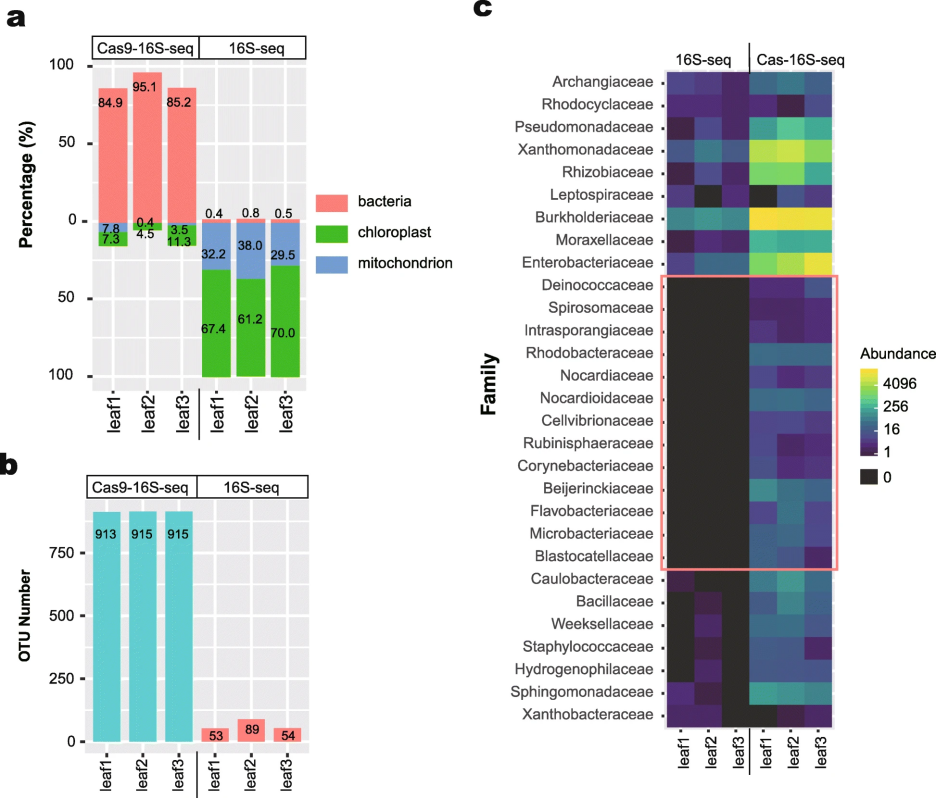
Cas-16S-seq与常规16S-seq分析水稻叶片样品的结果比较
在本研究中,谢卡斌课题组开发了名为Cas-16S-seq的方法,该方法利用Cas9核酸酶和特异的向导RNA(gRNA)切割16S-seq测序文库中的植物16S rRNA靶标,从而消除或减少宿主序列污染。该研究以前期CRISPR-Plant所使用的gRNA设计工具为基础,建立了一套生物信息学流程,通过比较植物与细菌的16S rRNA基因序列,设计了靶向水稻16S rRNA的高特异性gRNA。研究人员分别以人工混合样品、稻田土壤、水稻根系和水稻叶片样品为例,对Cas-16S-seq和常规16S-seq的数据进行了比较。
研究结果表明 Cas-16S-seq显着降低了水稻16S rRNA基因序列的比例:根系样品中,水稻的序列从63.2%降至2.9%;叶片样品中,从99.4%降至11.6%。因此与常规16S-seq相比,Cas-16S-seq能检测到的细菌信息有了大幅度的提高。我们的分析结果也表明Cas-16S-seq特异性高,没有检测到gRNA脱靶切割样品中细菌16S rRNA基因的现象,说明该方法不会引入人为偏差。
该研究解决了16S-seq分析植物样品时大量宿主序列污染的难题,为植物微生物组学研究提供了一个强有力的工具,也为优化微生物组学研究工具提供新思路。
植物科学技术学院2018级博士研究生宋露洋为论文第一作者,谢卡斌教授为论文通讯作者。本研究获得了国家自然科学基金、转基因新品种培育重大专项、作物遗传改良国家重点实验室和华中农业大学的资助。
审核人:谢卡斌
论文链接:https://microbiomejournal.biomedcentral.com/articles/10.1186/s40168-020-00859-0
