群体水平揭示玉米DNA甲基化变异对表型的贡献 | Genome Biology
论文标题:Population-level analysis reveals the widespread occurrence and phenotypic consequence of DNA methylation variation not tagged by genetic variation in maize
作者:Jing Xu, Guo Chen et al.
发表时间:2019/11/19
微信链接:点击此处阅读微信文章
DNA甲基化对植物的生长发育和环境响应起着十分重要的作用。作为一种较为稳定的遗传信息,DNA甲基化既可由DNA序列变异诱发,也可独立于DNA序列产生纯表观遗传变异,这种独立的程度和DNA甲基化对基因表达以及表型变异的影响一直是研究热点。近日,华中农业大学和美国明尼苏达大学合作解析了玉米自然群体中的DNA甲基化变异,揭示了DNA甲基化变异的遗传基础以及DNA甲基化变异与基因表达和表型的关联。该成果发表在开放获取期刊Genome Biology上。
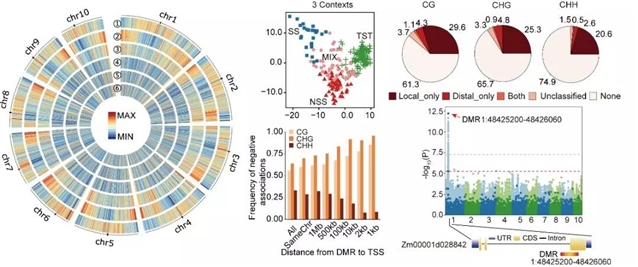
近年来,高通量测序技术和基因型检测技术的发展极大促进了作物遗传改良,育种家可以根据大量的SNP数据建立模型,预测表型和进行基因组选择,这一做法的前提是性状变异由DNA序列变异(SNP)决定。但是,对于很多性状,DNA序列变异通常只能解释部分的遗传力,仍有相当一部分遗传力无法被解释,许多学者将这种现象称为“消失的遗传力”,如何寻找并利用这部分遗传力是遗传学家和育种学家面临的一个重大挑战。表观遗传变异指不依赖于DNA序列的、可引起基因表达变化、并可遗传给后代的变异,包括DNA甲基化等染色质修饰。研究表明DNA甲基化在玉米不同基因型间存在巨大变异,但是,由于玉米基因组复杂和测序成本高,一直缺乏高通量低成本DNA甲基化检测技术,因而无法在群体水平研究DNA甲基化变异的程度及其生物学功能。
本研究中,研究人员拓展了前期开发的液相探针捕获技术(Li等, Nucleic Acids Research, 2015, 43:e81),包括了更多的DNA甲基化变异区段,这些区段的选择基于研究人员前期对玉米基因组DNA甲基化的研究成果,包括了基因组中最有可能发生DNA甲基化变异、影响基因表达或产生表型、同时又能被有效监测的区段。基于这一技术,研究人员对263份来自热带、亚热带和温带的玉米自交系的DNA甲基化进行了分析,寻找到了大量DNA甲基化差异区段(Differentially methylated region, DMR),结合高密度SNP数据、叶片和籽粒基因表达数据以及籽粒代谢数据,解析了DNA甲基化变异的遗传基础及其对表型变异的影响。研究发现DNA甲基化变异广泛存在,且绝大多数变异(>60%)不与DNA序列变异关联,同时发现DNA甲基化变异对基因表达的影响依赖于组织和序列环境。此外,DMR与代谢性状的关联分析发现约16%的性状与DMR显著关联,有趣的是,与性状关联的DMR中超过半数不与SNP关联,且有些性状只与DMR关联而不与SNP关联,提示DNA甲基化携带了特有的遗传信息。
本研究系统解析了DNA甲基化变异的遗传基础,并证明DNA甲基化可调控基因表达和表型,为解释消失的遗传力提供了新的支持。这是第一次在群体水平分析作物中DNA甲基化的变异和功能。
华中农业大学李青教授和明尼苏达大学Nathan Springer教授为论文的共同通讯作者;博士生徐静和陈果为共同第一作者;华中农业大学严建兵教授和李林教授参与了相关研究工作。该研究得到国家重点研发计划、华中农业大学科研启动经费和华中农业大学自主科技创新基金等的资助。
摘要:
Background
DNA methylation can provide a source of heritable information that is sometimes entirely uncoupled from genetic variation. However, the extent of this uncoupling and the roles of DNA methylation in shaping diversity of both gene expression and phenotypes are hotly debated. Here, we investigate the genetic basis and biological functions of DNA methylation at a population scale in maize.
Results
We perform targeted DNA methylation profiling for a diverse panel of 263 maize inbred genotypes. All genotypes show similar levels of DNA methylation globally, highlighting the importance of DNA methylation in maize development. Nevertheless, we identify more than 16,000 differentially methylated regions (DMRs) that are distributed across the 10 maize chromosomes. Genome-wide association analysis with high-density genetic markers reveals that over 60% of the DMRs are not tagged by SNPs, suggesting the presence of unique information in DMRs. Strong associations between DMRs and the expression of many genes are identified in both the leaf and kernel tissues, pointing to the biological significance of methylation variation. Association analysis with 986 metabolic traits suggests that DNA methylation is associated with phenotypic variation of 156 traits. There are some traits that only show significant associations with DMRs and not with SNPs.
Conclusions
These results suggest that DNA methylation can provide unique information to explain phenotypic variation in maize.
(来源:科学网)
