科学家在人类肠道菌群中鉴别出6000多个抗生素耐药基因
近日,一项刊登在国际杂志Nature Microbiology上的研究报告中,来自伯明翰大学的科学家们通过研究利用一种创新性技术,在栖息于人类肠道中的细菌中鉴别出了数千个抗生素耐药基因。人类机体肠道中有数以万亿计的微生物,其中主要是细菌,大部分的细菌都对抗生素比较敏感,但人类肠道中也有一部分细菌会产生对抗生素耐受的机制,然而目前研究人员并没有深入研究理解能够介导肠道菌群对抗生素产生耐药性的相关基因。
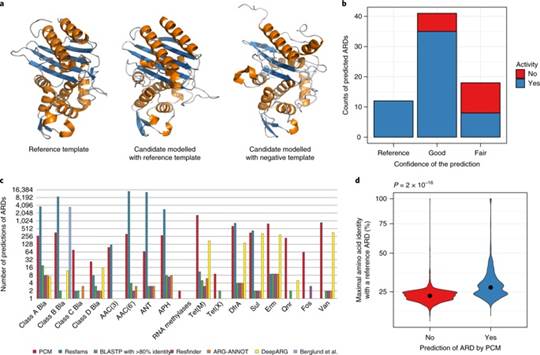
这项研究中,研究人员就开发了一种新方法,其能通过对比已知的抗生素耐药酶类和肠道菌群所产生的蛋白质的三维结构,来鉴别肠道菌群中的耐药性基因。借助这种方法,研究人员构建出了肠道中数百万个基因的目录,随后他们发现了6000多个抗生素耐药基因与之前在致病菌中发现的基因有很大的区别。
研究者Willem van Schaik教授说道,大部分的肠道菌群与人类宿主的关系都是无害的,然而肠道同样也是诱发机体感染的细菌喜欢的住所,很不幸的是,这些细菌如今开始对抗生素产生耐药性,因此我们就需要理解其诱发耐药性的分子机制。通过将已知抗生素耐药性蛋白的结构与人类肠道中细菌所产生的蛋白质的结构进行对比,研究人员在人类肠道中发现了数千个新型的抗生素耐药基因,这就强调了人类肠道环境中抗生素耐药基因的巨大多样性。
最后研究者指出,在所鉴别出的耐药性基因中,大部分基因都存在于一些与人类宿主之间处于无害关系的细菌中,因此其或许并不会对人类健康构成直接威胁;然而,如今抗生素的持续使用常常会导致很多耐药性基因转移到致病菌细胞中,这就会降低未来临床中使用抗生素治疗感染性疾病的有效性。(来源:生物谷Bioon.com)
Prediction of the intestinal resistome by a three-dimensional structure-based method
Abstract The intestinal microbiota is considered to be a major reservoir of antibiotic resistance determinants (ARDs) that could potentially be transferred to bacterial pathogens via mobile genetic elements. Yet, this assumption is poorly supported by empirical evidence due to the distant homologies between known ARDs (mostly from culturable bacteria) and ARDs from the intestinal microbiota. Consequently, an accurate census of intestinal ARDs (that is, the intestinal resistome) has not yet been fully determined. For this purpose, we developed and validated an annotation method (called pairwise comparative modelling) on the basis of a three-dimensional structure (homology comparative modelling), leading to the prediction of 6,095 ARDs in a catalogue of 3.9 million proteins from the human intestinal microbiota. We found that the majority of predicted ARDs (pdARDs) were distantly related to known ARDs (mean amino acid identity 29.8%) and found little evidence supporting their transfer between species. According to the composition of their resistome, we were able to cluster subjects from the MetaHIT cohort (n?=?663) into six resistotypes that were connected to the previously described enterotypes. Finally, we found that the relative abundance of pdARDs was positively associated with gene richness, but not when subjects were exposed to antibiotics. Altogether, our results indicate that the majority of intestinal microbiota ARDs can be considered intrinsic to the dominant commensal microbiota and that these genes are rarely shared with bacterial pathogens.
原文链接:https://www.nature.com/articles/s41564-018-0292-6
