南海海洋所利用生物合成技术获得强效抗结核抗生素
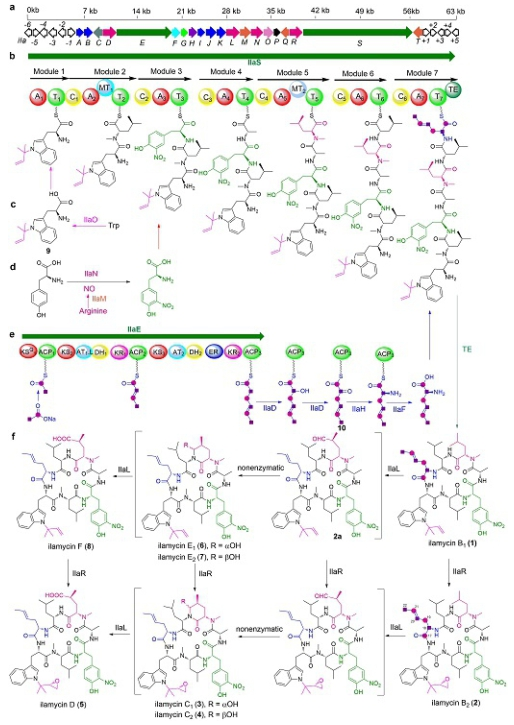
怡莱霉素的生物合成基因簇及其生物合成过程。(a)怡莱霉素的生物合成基因簇;(b)怡莱霉素的骨架结构的合成过程;(c)色氨酸的异戊烯基化过程;(d)酪氨酸的硝基化过程;(e)L-2- 氨基 -4- 己烯酸的生物合成机制;(f)怡莱霉素的氧化后修饰过程。
近日,中国科学院南海海洋研究所热带海洋生物资源与生态重点实验室,中科院广州生物医药健康研究院呼吸疾病国家重点实验室,以及广东医科大学合作的研究论文,以 Biosynthesis of ilamycins featuring unusual building blocks and engineered production of enhanced anti-tuberculosis agents 为题发表在 Nature Communications 上,报道了从深海放线菌中发现了具有抗结核杆菌系列活性物质,通过生物合成技术优化改造获得低细胞毒活性、强抗结核杆菌活性的化合物怡莱霉素 E。
结核病是最为严重的传染性疾病,死亡率已超过 HIV,位居传染性疾病之首。据 WHO 结核病年报报道,全球 2015 年新增结核病人 1040 万人,其中死亡约 180 万人。目前,临床上应用的 4 个一线抗结核药物和 8 个二线抗结核药物均是发现于上世纪的 40 年代到 70 年代,此后再无新的抗结核药物出现。直至 2012 和 2014 年,FDA 和欧盟先后批准贝达喹啉(Bedaquiline)、Delamanid 与其他抗结核药物联合使用,治疗多重耐药结核杆菌的感染,但存在副作用严重且价格昂贵问题,临床应用受到一定限制。多重耐药和泛耐药或全耐药结核菌株在迅速增长,加上结核病与 HIV 并发感染等情况的出现,对本已严峻的结核病防控形势带来了新挑战,因此对高效低毒的新型抗结核药的需求日益迫切。
海洋中蕴藏着丰富的海洋微生物资源,其代谢产物尚未被充分发掘利用。深海环境营养匮乏,生活在深海的微生物为了争夺生存空间,会产生抗生素抑制周边微生物的生长。研究人员利用这一化学生态学原理从深海微生物中筛选抗菌活性物质。经过筛选,他们发现一株深海(-3560 米)来源的放线菌发酵提取物对抗耻垢分枝杆菌具有较强的选择性抑制活性,并对肿瘤细胞增殖具有一定的抑制活性。研究人员对菌株进行扩大培养和活性追踪分离,利用多种波谱技术解析了 6 个命名为怡莱霉素的活性物质结构(图 1,化合物 1 -6),并用单晶衍射技术测定了 3 个化合物的绝对构型。怡莱霉素属于环七肽类化合物,结构中含有独特的 l -2- 氨基 -4- 己烯酸,3- 硝基 -l- 酪氨酸,和异戊烯基化的色氨酸结构单元;其中,l-2- 氨基 -4- 己烯酸结构单元在其它天然产物中未见报道。此外,怡莱霉素结构中的亮氨酸结构单元和异戊烯基均被氧化。
科研人员阐明怡莱霉素的生物合成机制,并利用生物工程技术构建突变株,期望构建新结构衍生物,简化代谢产物组分,并提高目标组分的产量。科研人员对怡莱霉素产生菌进行了基因组测序,根据怡莱霉素的结构特征,通过生物信息分析和基因敲除技术,确定了负责怡莱霉素的生物合成基因簇(图 1,a)和骨架结构的生物合成过程(图 1,b)。对四个功能基因进行敲除和代谢产物分析,结合前体分子喂养及同位素标记实验,阐明了新颖的 I 型聚酮合酶 IlaE、细胞色素 P450 氧化酶 IlaD、氨基转移酶 IlaH 和 II 型硫酯酶 IlaF 负责稀有结构单元 l -2- 氨基 -4- 己烯酸的生物合成(图 1,e)。对怡莱霉素生物合成基因簇中的一氧化氮合酶 IlaM 和细胞色素 P450 氧化酶 IlaN 所编码的基因进行敲除和代谢产物分析,及前体分子回补实验,阐明了 L - 酪氨酸的硝基化过程(图 1,d)。通过对怡莱霉素生物合成基因簇中的异戊烯基转移酶 IlaO 和另外两个细胞色素 P450(IlaL 和 IlaR)所编码的基因进行敲除,代谢产物分析,以及中间体的分离和结构鉴定,阐明了 IlaO 在怡莱霉素形成过程中负责色氨酸的异物烯基化的前修饰作用(图 1,c),IlaL 和 IlaR 这两个酶在怡莱霉素生物合成过程中分别负责亮氨酸末端甲基的羧基化及异物烯基的环氧化的后修饰(图 1,f)。通过上述研究,科研人员发现,怡莱霉素生物合成过程中的三个前修饰和两个后修饰过程,并构建高产菌株,定向生产新结构衍生物怡莱霉素 E 和 F。
科研人员系统评价了分离和工程改造获得的系列怡莱霉素衍生物对结核分枝杆菌 M. Tuberculosis H37Rv,5 种人体肿瘤细胞株和 2 种正常细胞株的抑制活性,研究发现,怡莱霉素 E 的体外抗结核活性为 9.8 nM,是一线抗结核药物利福平活性的 30 倍,怡莱霉素 E 对乳腺癌等肿瘤细胞也显示出一定抑制活性,对正常细胞的毒性较低,在抗结核活性和细胞毒性之间的选择性指数为 400-1500,显示出较好的安全性窗口,具有成药潜力。该研究阐明了海洋微生物复杂活性代谢产物怡莱霉素的生物合成机制,并获得具有更好活性和低毒性的抗结核活性化合物,为新型抗结核药物的进一步开发提供了化学实体。
研究工作得到了国家自然科学基金、中科院、广东省自然科学基金团队和广州市科技计划项目的资助。(来源:中国科学院南海海洋研究所)
Biosynthesis of ilamycins featuring unusual building blocks and engineered production of enhanced anti-tuberculosis agents
Abstract Tuberculosis remains one of the world’s deadliest communicable diseases, novel anti-tuberculosis agents are urgently needed due to severe drug resistance and the co-epidemic of tuberculosis/human immunodeficiency virus. Here, we show the isolation of six anti-mycobacterial ilamycin congeners (1–6) bearing rare L-3-nitro-tyrosine and L-2-amino-4-hexenoic acid structural units from the deep sea-derived Streptomyces atratus SCSIO ZH16. The biosynthesis of the rare L-3-nitrotyrosine and L-2-amino-4-hexenoic acid units as well as three pre-tailoring and two post-tailoring steps are probed in the ilamycin biosynthetic machinery through a series of gene inactivation, precursor chemical complementation, isotope-labeled precursor feeding experiments, as well as structural elucidation of three intermediates (6–8) from the respective mutants. Most impressively, ilamycins E1/E2, which are produced in high titers by a genetically engineered mutant strain, show very potent anti-tuberculosis activity with an minimum inhibitory concentration value ≈9.8 nM to Mycobacterium tuberculosis H37Rv constituting extremely potent and exciting anti-tuberculosis drug leads.
原文链接:http://www.nature.com/articles/s41467-017-00419-5
