高通量测序(NGS)的这10年

随着人类基因组计划(human genome project )在2003年顺利完成,基因组测序技术取得了长足的进步,这直接导致了每兆基因组成本的大幅下降以及检测的基因组数量越来越多。人们对基因组的复杂性深感震惊,这也引导着测序技术的进一步发展。最近的一些突破性技术使得测序技术在更短的时间内可以获得更多的数据量。与之对应的是,还有一些技术的进步使得单条序列的测序读长变得更长——这对解析结构性的复合区段是极其必要的。这些进展给科研人员以及医疗诊断人员提供了一个绝佳的平台使得人们对基因组变异导致的表型变化以及疾病发生有了进一步的了解。
近日,美国冷泉港实验室联合加州大学戴维斯分校的研究人员在国际著名评论型综述杂志Nature Reviews Genetics(影响因子41)上发表了一篇评论型综述。该综述对高通量测序的技术原理以及各平台的优势比较和实践应用进行了深入浅出的分析。
介绍
自从DNA的双螺旋结构被人们解析开始1,人们在探究健康与疾病的基因组的复杂性与差异性上做出了巨大的努力。为了支持人类基因组计划的顺利进行2,人们在仪器和试剂上做出了巨大的改进。该计划的完成使得人们强烈的意识到人们需要更多更好的技术与数据分析能力来回答随之而来的一系列生物学问题。然而,通量的限制以及居高不下的测序成本成为了人们进一步了解基因组的一道坎。2000年之后推出的高通量测序平台很好地解决了这个问题,人类基因组测序的成本直接因此下降50000倍,并且由此产生了一个新的名词:下一代测序(next-generation sequencing,NGS)3。在过去的十年中,NGS技术不停的在进步——测序的数据量增加了100-1000倍4。这些技术上的进展使得人们甚至可以在一条read上读出整条基因组序列。根据Veritas Genomics的数据5,人类基因组测序的成本也已经下降到1000美元/人。不仅如此,该技术已经广泛在临床诊断上得到应用3,6。
但是,尽管NGS技术非常重要,却并非完美。与NGS技术一道出现的是该技术带来的一系列问题。NGS可以提供海量的数据量,但是其质量却有待提高(有报道,NGS在序列拼接过程中,错误率在0.1-15%范围内),并且NGS的序列读长普遍较低(每条read的长度在35-700bp之内7,这比普通的Sanger测序要短),这意味着需要更严格复杂的序列拼接。尽管长读长测序可以克服NGS的这一大弱点,但相对而言,成本较高并且通量较低,这也限制了该技术的进一步应用。最后,NGS同时还和其他的技术之间存在着竞争的关系。
短读长(read)的NGS测序
测序模版克隆法生成综述
短读长测序方法包含两种:边连接边测序(sequencing by ligation, SBL)以及边合成边测序(sequencing by synthesis, SBS)。在SBL方法中,带有荧光基团的探针与DNA片段杂交并且与临近的寡核糖核酸连接从而得以成像。人们通过荧光基团的发射波长来判断碱基或者其互补碱基的序列。SBS方法通常使用聚合酶,而且,诸如荧光基团在链的延伸过程中被插入其中。绝大多数的SBL和SBS方法,DNA都是在一个固体的表面上被克隆。一个特定区域内成千上万个拷贝的DNA分子可以增加信号和背景信号的区分度。大量的平行同样对上百万的reads的读取大有帮助,每个平行只有唯一的DNA模板。一个测序平台可以同时从上百万的类似反应中读取数据,因此可以同时对上百万的DNA分子进行测序。
产生模板的克隆有几个方法:基于磁珠(bead-based),固相介质(solid-state)以及DNA微球技术(DNA nanoball)(图1)。DNA模板产生的第一步就是样本DNA的片段化,接着是连接到一个为了克隆和测序而设计的接头上。在磁珠法的准备过程中,一个接头和寡核糖核酸片段互补并且固定在珠子上(图1a)。DNA模板通过使用油包水PCR(emulsion PCR,emPCR)8得以扩增。单个珠子上被克隆得到的DNA片段可以达到上百万个9。这些珠子可以被分为glass surface10或者PicoTiterPlate(罗氏诊断)11。固相介质扩增12避免了油包水PCR,取而代之的是在固相介质上直接进行PCR13(图1b,c)。该方法中,正向和反向引物结合在芯片的表面,这些引物给单链DNA(single-stranded DNA,ssDNA)提供了末端的互补序列供其结合。最近,几个NGS的平台都是用了模块化的flow cells。
BGI使用的Complete Genomics technology测序技术是唯一一个在溶液中完成模板富集的技术。在这种情况下,DNA被多次连接,成环以及剪切从而为了产生一个包含4个不同接头的环状的模板。通过旋转环状扩增(rolling circle amplification,RCA),可以最多产生超过200亿的DNA微球(图1d)。微球混合物随后被分配到芯片表面上,使得每个微球可以占据芯片的一个位点14。
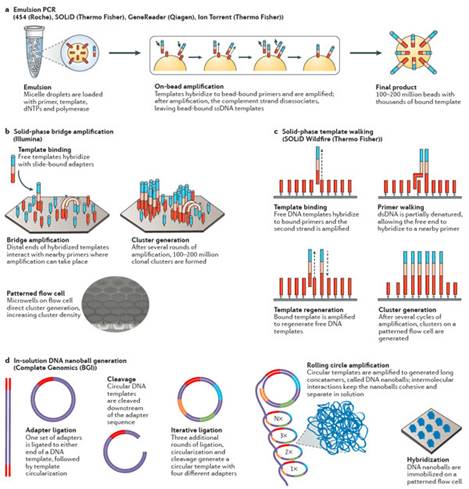
边连接边测序(SOLiD和Complete Genomics)
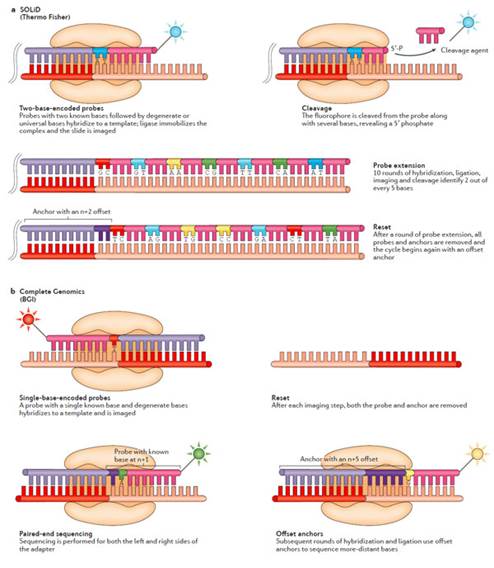
从根本上来说,SBL法包含了杂交和对标记的探针的连接15。探针包含了一到两个特定碱基序列和一系列通用序列,这可以使得探针与模板之间进行互补配对。锚定的片段则包含一段已知的和接头互补的序列用于提供连接位点。连接之后,模板被系统进行测序反应16。在锚和探针复合物或者荧光基团被完全移除之后,也或者连接位点重新生成之后,新的循环又重新开始了。
SOLiD平台使用的是双碱基编码的探针,每个荧光基团信号代表了一个二核糖核酸17。因此,原始输出的数据并非直接和已知的核糖核酸相连。因为有16种可能的二核糖核酸组合并不能单独结合荧光基团。每四种组合使用一种荧光信号,共有四种荧光信号。所以,每种连接信号代表了几种可能的二核糖核酸组合。SOLiD测序过程由一系列的探针-锚的结合,连接,图像获取以及切割的循环组成。
Complete Genomics使用探针-锚的连接方式(cPAL)或者探针-锚的合成方式(cPAS)来进行测序14。在cPAL中(图2b),锚的序列(与四种接头序列其中之一的互补)以及探针杂交到DNA微球的不同位置。每个循环中,杂交探针是一组特定位置已知碱基序列的探针的一员。每个探针包涵一段已知序列的碱基以及对应的荧光基团。获取图像之后,全部的探针-锚复合物被移除,新的探针-锚复合物被杂交。cPAS方法是cPAL的修改版,增加了read的长度;然而,目前来说,该方法还是有局限性的。
边合成边测序(Sequencing-by-synthesis)
SBS的方法是指那些依赖于大量的DNA聚合酶来进行测序的方法。但是,SBS中依然包括了各种不同的测序原理。本文中,SBS方法被分为循环可逆终止(Cyclic reversible termination, CRT)以及单核糖核酸增加(single-nucleotide addition, SNA)18。
边合成边测序:CRT(Illumina,Qiagen)
CRT方法是根据类似于Sanger测序的终止反应来界定的,其3\&\#39\;-OH基团被屏蔽而被阻止继续延伸19,20。在反应开始时,DNA模板被一段和探针序列互补的接头结合,DNA聚合酶也是从这段序列开始结合。每个循环过程中,四种单独标记的复合物和3\&\#39\;屏蔽的脱氧核糖核酸被添加进反应中。在延伸过程中每结合一个dNTP,其他没有被结合的dNTPs被移除,并且获取图像来确定是那个碱基在某个簇中被结合。荧光基团以及屏蔽基团随后被移除并且开始一轮新的反应。
Illumina的CRT和其他平台相比,代表了最大的测序平台市场。Illumina短读长测序的设备可以从台式的低通量单位到大型的超高通量,如应用于全基因组关联分析(whole-genome sequencing,WGS)。dNTPs是通过两个或者四个激光通道来对荧光进行分析的。在绝大多数Illumina平台上,每种dNTP结合一种荧光基团,因此需要四种不同的激光通道。而NextSeq和Mini-Seq则使用的是双荧光基团系统。
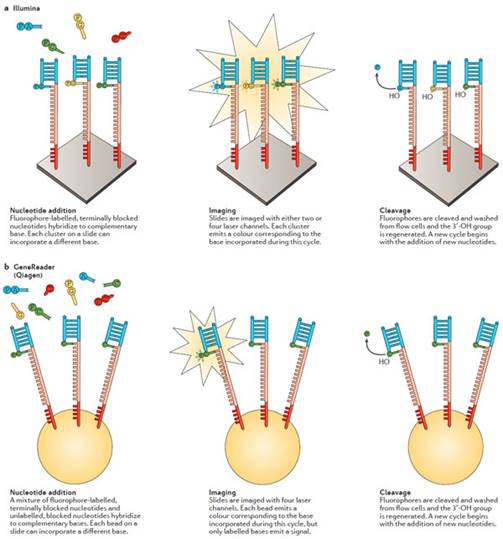
2012年,Qiagen获得了Intelligent BioSystems CRT平台,并且在2015年将该平台命名为GeneReader重新推出并且使之商业化22(图3b)。与其他平台不同的是,该平台打算做一站式的NGS平台,从样本制备到数据分析,全部一站式解决。为此,GeneReader系统整合了QIAcube样本制备系统和Qiagen Clinical Insight平台用于不同的数据分析。GeneReader平台的技术原理与Illumina平台基本一致。然而,该平台并非让每个DNA模板都去结合带有荧光基团的dNTPs23,而是只要足够的dNTPs结合到模板上就可以完成鉴定。
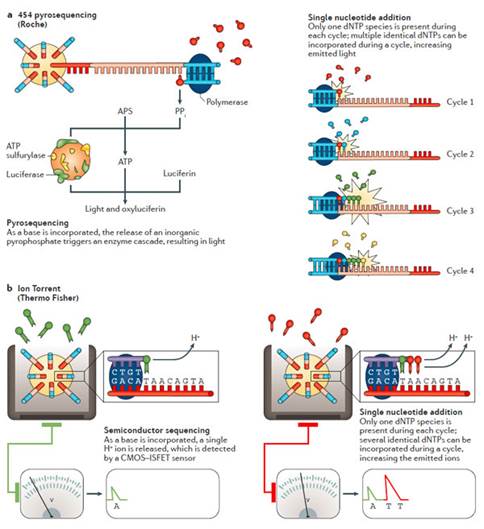
边合成边测序:SNA(454,Ion Torrent)
与CRT不同的是,SNA方法依赖于单信号标记dNTP来对链进行延伸。四种核糖核酸都必须反复添加到测序反应过程中。不仅如此,SNA不需要将dNTP屏蔽,因为测序反应过程中下一个碱基的缺失会阻止链的延伸。碱基的寡聚体则是一个例外,在这种情况下,信号的强度会随着dNTP数量的增加而成比例的增强。
第一个NGS仪器是454焦磷酸测序仪24。这种SNA系统将结合有模板的珠子以及酶混合物分配到PicoTiterPlate中。由于一个dNTP只能结合到一条链上,酶复合物会对其产生生物荧光。一个特定的珠子中的一个或多个dNTPs可以通过电荷共轭偶联设备(charge-coupled device, CCD)检测到的荧光来确认(图4a)。
Ion Torrent是第一个没有光学感应的NGS平台25。与酶化学复合物产生的信号相比,Ion Torrent平台检测的是dNTP中释放出来的H离子。pH值的改变通过(integrated complementary metal-oxide-semiconductor,CMOS)以及(ion-sensitive field-effect transistor,ISFET)来检测(图4b)。传感器对pH的变化对于连续碱基的检测还不够完善,因此在测量同一碱基连续出现时的数量可能会有所误差。
短读长平台的比较
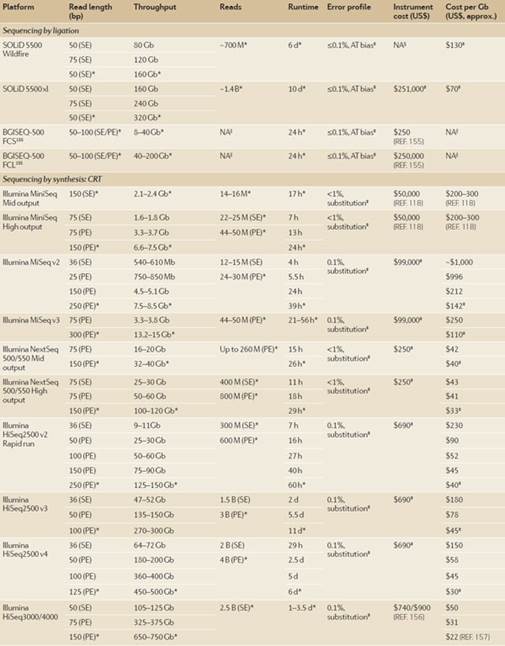
每个平台在通量,成本,错误率以及read结构上都大相径庭(表一)。尽管有多家NGS技术供应商,NGS研究最常用的还是Illumina平台21。尽管该平台极为稳定,数据可靠,但是基于其使用的单一测序的方法26-28,既然具有系统偏好性的问题。因此,新技术的发展使得研究人员能够有完整的测序方案来获得完整的序列信息。
SOLiD与Complete Genomics系统使用的SBL技术准确率非常高(~99.999%)7,14,因为每个碱基都会被标记多次。虽然这些技术非常准确,但是在敏感性与特异性之间依然不能达到完美的平衡,当一些错误的碱基变化出现时,真实的碱基变化可能被忽略29-31。该类技术在应用上最大的限制可能就是其过短的读长。尽管所有的平台都能产生单末端和双末端的reads,SOLiD的最大读长只能达到75bp,Complete Genomics只能达到28-100bp33,使得其在基因组拼接和结构变异研究中的可操作性大大降低。不幸的是,SOLiD系统不仅受制于运行时间,还受制于其工业生产。另外,尽管cPAL计划准备在成本和通量上和Illumina竞争,却在2016年被迫下马,该技术仅在人类WGS中有所应用33,34。cPAS的BGISEQ-500系统则受制于中国大陆政府。
Illumina由于其技术成熟,平台之间高度互补性与交叉性,使得其在短读长测序上大占优势。Illumina的产品覆盖了从低通量的Mini-Seq到超高通量的HiSeq X系列,其中HiSeq X系列最多可以在一年内产生1800多个30×覆盖度的人类基因组数据量。此外,其运行时间,read结构以及read长度(最大300bp)都在不停的改进。但是,作为一个依赖于CRT技术的Illumina平台,相对于SNA平台的优势在于其在读取核糖核酸多聚体(同一种核糖核酸多次出现)时较低的错误率。尽管SNA平台总体上的准确率可以达到99.5%35,但是在读取那些高AT富集或者高GC富集的片段的时候错误率差强人意32,37,38。在2008年,据Bentley等报道,Illumina平台鉴定到的人类单核糖核酸多态性(SNPs)与基因芯片鉴定的SNPs具有惊人的一致性35。但是,这种高度的敏感性也随之带来了2.5%左右的错误率。因此,其他小组计划使用Sanger测序来对鉴定到的SNPs进行重新测序以便区分测序错误导致的SNPs与真实的基因突变导致的SNPs35,39,40。在对所有的可能性都进行优化之后,Illumina平台被大量的研究人员认可,在大量的领域中均有涉及:WGS的基因组测序与外显子测序;遗传学应用如染色质免疫共沉淀——测序(chromatin immunoprecipitation followed by sequencing)41;ATAC-Seq(transposase-accessible chromatin using sequencing)42或者DNA甲基化测序(Methyl-Seq)43;RNA转录组测序(transcriptomics applications through RNA sequencing, RNA-seq)44等等。NextSeq与MiniDeq平台使用的双色标记系统通过降低双色通道的扫描与荧光基团的使用达到成本并且增加测序速度。然而,双通道系统却会略微增加测序的错误率45。HiSeq X是目前最高通量的仪器,但其由于通量过大,因此只在部分应用上得以使用,如WGS与全基因组甲基化测序。不仅如此,HiSeq X更大的局限在于其高昂的成本,以至于超过了绝大多数单位的可接受程度。
Qiagen的GeneReader是专为临床诊断设计的,其主要关注点在肿瘤基因panels46上,也因此其局限性较大。根据对其运行时间与功能的分析,GeneReader与Illumina的MiSeq较为相似46。尽管还没有使用数据,但是GeneReader和MiSeq平台有相同的优缺点。
454平台和Ion Torrent平台相比于其他的短读长平台而言,能够提供较长的read读长,分别大约在700bp与400bp,因此在基因组结构较为复杂的研究上应用较多。然而,由于同样都是基于SNA技术,它们都拥有相同的缺点。虽然,其在非碱基多聚体(non-homopolymer)的测序上正确率与其它NGS平台相差无几,但其插入与缺失(Insertion and deletion,indel)是最大的问题。同一碱基的多聚体是该类技术最大的问题所在。有报道,对同一碱基的多聚体的测序误差能够达到6-8个碱基之多47,48。不幸的是,尽管Ion Torrent依然在紧跟快速进化的NGS平台的步伐,454平台却由于成本与应用范围过于狭小却已经被罗氏公司停产。
Ion Torrent平台为不同的研究人员的不同需求提供了不同的芯片与设备,通量从50Mb到15Gb不等,运行时间也从2小时到7小时不等。这一点使得其几乎是所有目前的二代测序平台中最快的一个。这也使得其在基因panel与精准临床诊断上大有优势50,包括转录组与可变剪切鉴定51。Ion Torrent先后发布Ion Personal Genome Machine (PGM) Dx与Ion S5系列希望于在临床诊断上打开疆土。与Ion Chef文库制备试剂盒和芯片上样设备结合使用,S5系列希望能够成为最方便操作的设备,消除其它Ion Torrent设备对氩的依赖。但是,其最大的缺点在于Ion PGM Dx系统可以进行双向测序,更高通量的Ion Proton与S5系统却并不支持双向测序,也因此限制了其在大范围基因组测序与转录组结构上的应用。
长读长(read)的NGS测序
综述
基因组是一个复杂的复合物,其中包含了多种重复序列,拷贝数变化,结构变异。这些与进化,适应以及疾病密切相关54-56。然而,许多复合物元件由于过长,导致短读长测序并不能够完美的对其进行测序。长读长测序的reads可以达到几千个碱基,这使得可以对大的结构进行功能解析。此类的长读长测序产生的单一长序列可以跨越复合物或者重复序列。长读长测序在转录组测序过程中也大有益处,因为长读长的reads可以跨越完整的mRNA的转录本而不需要拼接。这可以使得研究人员可以鉴定到更多的基因亚型等。
最近,人们开发出了两种长读长测序的实验方案,分别是:单分子实时测序(single-molecule real-time sequencing )以及依赖于已有短读长技术体外构建长读长的合成法。单分子法与短读长测序完全不同,因为单分子法不需要对模板进行扩增来产生足够测序仪读取的信号,也不需要轮番添加dNTP。而合成法并非产生原始的长读长的reads,而是通过利用barcodes来进行拼接获得长片段。
表一:NGS平台概述。
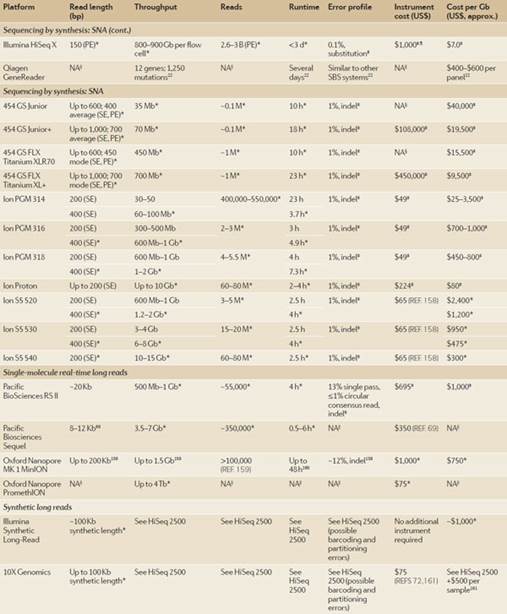
单分子长读长测序(PacBio和ONT)
最近这段时间,最常用的长读长测序法平台就是使用PacBio Biosciences(PacBio)57的单分子实时测序法(single-molecule real-time sequencing, SMRT)(图5a)。该设备使用了一个特制的流动单元,其中包含了成千上万的单独的底部透明的皮升孔(picolitre wells)——zero-mode waveguides(ZMW)58。短读长SBS技术需要使得聚合酶结合DNA,沿着DNA进行扩增,而PacBio则固定聚合酶在空的底部,让DNA链通过ZMW。由于有聚合酶有固定的位置,因此该系统可以对单分子DNA进行测序。dNTP结合在每个孔的单分子模板上,通过激光或者成像设备记录ZMW底部标记在核糖核酸上的发射波长的颜色与持续时间来进行序列的读取。聚合酶在结合dNTPs的过程中,切割dNTP结合的荧光基团,使得荧光基团在第二个标记的碱基进入ZMW前将前一个荧光基团去除。SMRT平台也使用了独特的环状模板,这种方式的模板可以使得聚合酶反复读取模板的序列。尽管这种方法不太容易对长度大于3kb的片段反复读取,但是短的模板却可以反复读取多次57,59。由于多次读取同一序列,因此系统会产生多次测序后的保守序列(consensus sequence, CCS)。
2014年,第一个消费级别的nanopore测序仪的原型机——MinION在Oxford Nanopore Technologies(ONT)诞生。与其他平台不同的是,nanopore测序仪并不监测模板DNA结合或杂交的核糖核酸。其它平台通过监测次级信号,光,颜色或pH等来进行碱基序列的读取,二nanopore则直接对天然的ssDNA分子进行读取。为达成此,DNA需要通过一个蛋白孔(protein pore)(图5b),孔也会因为DNA分子的通过导致的电压阻塞(voltage blockade)的发生。对这些电荷瞬时的追踪称为squiggle space,特定DNA序列通过孔会产生特定的电压改变,这被称为k-mer。相比于1-4种可能的信号,nanopore拥有1000多种可能的k-mer,尤其是当天然DNA序列中存在修饰的碱基的时候。最近的MK1 MinION流动单元由特殊应用的芯片组成,包涵了512个独立的通道,每秒可以读取70bp长度,到2016年预计能够增加到500bp/秒。新推出的PromethION设备是包含了48个独立流动单元的高通量平台。该项工作最多可以在2天内输出~2-4Tb的数据量,这使可能其成为HiSeq X系列的强力竞争者。与PacBio的环状模板类似的是,ONT MinION使用一个leader-harpin library结构。这使得正向DNA链可以通过孔,接着harpin蛋白结合双链,最后是反义链。这产生了1D和2D reads,1D链可以通过比对产生一个保守的2D read。
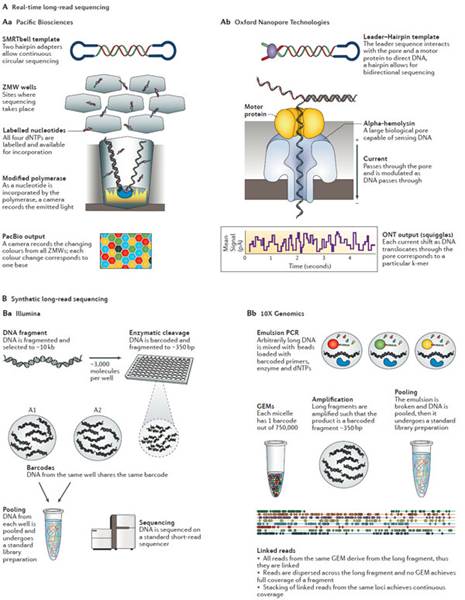
图5: 长读长实时测序原理。
长reads的合成
与真正测序的平台不同的是,合成长读长技术依赖于一个barcode系统来结合不同的片段,通过已有的短读长测序仪来获得长读长reads61。该方法将大的DNA分子分割成若干个小片段到微孔中或者乳液中。每个微孔或者乳液中的模板被切割并且加上了barcodes。这种方法允许在短读长测序仪上使用,测序后数据被通过barcode分开按照barcodes的序列进行拼接62。
合成法有两个系统:Illumina长片段合成平台(图5c)与10X Genomics乳液系统(图5d)。Illumina系统(Moleculo)分割DNA到小板上而不需要特殊仪器。然而,10X Genomics乳液系统(GemCode与Chromium)使用乳液分隔DNA并且需要微流体平台(microfluidic instrument)来进行测序前的准备工作。在其实浓度低至1ng的情况下,10X Genomics乳液系统可以任意切割长的DNA片段(最大达到100kb)到微粒(GEM)中,这种威力一般包含了≤0.3× 的基因组以及一个独特的barcode。
单分子测序与合成法测序的比较
人们对长读长测序越来越感兴趣,每个系统都有其优劣(表一)。最近长读长测序最受欢迎的是PacBioRS II。该设备可以产生超过50kb长度的单个read,长链建库测序平均长度为10-15kb。这种特性使得在基因组拼接与大范围基因组结构的应用中大有好处63,64。但是,长链的单个碱基错误率在15%左右65,使得人们对该仪器的使用有所顾虑66。不幸的是,这些错误随机分布每个reads,也因此必须有足够高的覆盖度来消除单个碱基错误率的负面影响67。PacBio的环状模板有时候也会出现错误。单个碱基测序次数越多,结果就越可靠,其最高准确率达到99.999%59,68。其高准确率与Sanger测序相似,使得该方法与Sanger测序一道成为SNPs的研究方法65。该设备的运行时间与通量受测序读长的影响,长的模板需要更长的时间。举例来说,1kb的库运行1小时测序每个分子可以产生7500个碱基,平均大约重复8次;而运行4小时每个分子可以产生大约30000个碱基(大约重复30次)。相反的是,10kb的库运行4小时产生30000个碱基只能重复3次左右。通量的限制以及高企的成本(1000美元/G),加上较高的覆盖度使得PacBio RS II成为那些较小的实验室难以应用的技术。然而,考虑到这些问题,PacBio推出了Sequel系统,其通量与RS II相比高出了7倍,使得30×覆盖度的人类基因组测序成本大幅下降一半69。
ONT MinION是一个小的(~3 cm× 10 cm)USB设备,并且可以在个人电脑上运行,使得其成为最小的测序平台。这使得MinION具有极高的便携性,并且在临床诊断中以及那些不容易到达的地方有着广泛的应用前景。尽管周边设备依然只有在实验室中才有,如文库准备的恒温器,这依然可以大幅减少设备空间。与其他平台不同,MinION在片段大小上是有限制的。理论上来讲,任意大小的DNA分子都可以在该设备上测序,但是实际情况是在对长片段进行测序过程中,是有所制约的70。作为ONT技术本身的特性,ONT拥有超过1000种独立的信号,这使得ONT拥有巨大的错误率——1D read大约在30%左右(主要是indel)。有效的对核糖核酸复合物的测序也是ONT MinION面临的一大问题。当核糖核酸复合物超过k-mer长度,就很难准确鉴定前一个k-mer何时离开孔而下一个k-mer何时进入孔。因为修饰的碱基会改变原有的k-mer设定的电压变化,所以碱基的修饰对MinION而言同样也是一大挑战。幸运的是,最近的一系列的对试剂以及算法的改进使得其准确率提高不少71。
应用
WGS正在成为NGS中最广泛的应用。通过该技术并且结合生物学应用,研究人员可以获得基因组信息中最值得注意的信息73。举例来说,2012年,Ellis等报道了基因与乳腺癌患者芳香酶抑制剂(aromatase inhibitor)治疗法之间的关联。他们指出突变,后果与诊断之间的关联,同样还有癌症相关基因的突变的富集。这提供了一个可能性,即:乳腺癌有不同的突变造成不同的表型,具有复杂的病理学75。最近的NGS平台的改进使得研究人员发现了一些几年前难以想象的新观点与机会。在2010年,1000基因组计划(1000 genomes project)开放了其从179个个体中获得的WGS原始数据以及697个个体的测序数据76。到2015年,研究人员已经构建了26个不同人群的2504个人的基因组群体77,78。给人们从种群的角度来观察人类的变异。但这还不是该项目的终点,越来越多的人的基因组正在被得以测序79-81。种群水平的测序已经成为人们更好的理解人类疾病的一个重要的工具,同样也得到了意想不到的结果。一个例子是,Sidore等82对2120个撒丁岛人(Sardinians)的WGS研究发现了一些新的和脂肪相关的基因以及炎症的标志物,给人们对血液胆固醇的分子机制的研究提供了新思路。
全外显子组测序(Whole-exome and targeted sequencing)83同样也广泛应用于测序的研究中。受制于基因组材料大小的局限,很更多的个人样本可以在一个测序中实现,增加了基因组研究的宽度以及深度。使用外显子测序,Iossifov84等对超过2500个单一的家庭进行测序,每个家庭都有一个小孩患有自闭症(autism spectrum disorder, ASD)。研究人员在30%的样本中发现了错意突变(missense mutations),基因干扰的突变(gene-disrupting mutations)以及拷贝数的变异。该工作与其他的工作一道鉴定到了ASD相关的基因突变85,86。其他证据表明,高覆盖度的WGS也可以解决复杂的变异以及临床样本的分析。2015年,Griffith等认为可以使用一个完美的跨平台的方法(包含靶向测序)来鉴定肿瘤中高可信度的SNPs。该方法中,作者认为10000×的覆盖度可以鉴定到稀有突变。由于10000×的覆盖度对于WGS而言实在过高,靶向测序便在临床中得到了广泛的应用。
NGS同样在基因的调控研究中有广泛的应用。蛋白-DNA互作可以通过染色质免疫共沉淀结合NGS测序(ChIP-seq)来得以研究41。利用NGS对修饰碱基的研究也是可行的。举例来说,甲基化测序包含了甲基化DNA的捕获与富集88,对甲基化与非甲基化区段的选择性消化89,90,91。但是,尽管利用此方法获得了很多重大的发现,修饰与捕获过程成为其最大的限制。2010年,Flusberg等92发表了一个概念性的研究方法,即:使用PacBio来区分甲基化与非甲基化的碱基。由于聚合酶即便是甲基化的碱基也能够延伸,但在甲基化位点上会停留更多的时间,因此这里改变的信号可以认为含有甲基化修饰。与之相同的是,nanopore平台也能够监测修饰的碱基,因为甲基化同样会影响鉴定到的电压的变化。这使得甲基化的测序可以在不需要化学操作的条件下进行93。
一个最近的NGS的范例是对长链DNA的测序。重复序列以及复合序列长久以来较难以拼接,短读长测序很难解决这个问题94-96。最近,Chaisson等97对长读长测序的使用使得其能够在人类GRCh37数据库中提交超过1Mb的新的序列,这些序列弥补甚至跨越了曾经的沟。Chaisson等还鉴定到了大于26000个超过50bp的indels,也因此,GRCh37数据库成为最有参考价值的几个基因组之一。除了简单的增加基因组数据可靠性之外,长读长还能够提供更有效的临床诊断98-100。
在对转录水平上的研究也因为NGS受益匪浅。今天,研究人员甚至能够使用NGS的深度测序对单个转录本进行研究。2014年,Treutlein等101使用了组织发育过程中不同细胞类群的单细胞RNA测序发现了用于鉴定细胞亚群的标志物。尽管长读长测序相对而言在对转录本的定量上不占优势,但是,长读长可以在研究转录组的结构上有所帮助51。举例来说,最近的人类长读长转录组测序研究表明 >10%的reads是新的可变剪切体102。
NGS最新的设备——nanopore测序仪,依然在寻找其定位的过程中。然而,研究人员正在将其快速的文库制备,实时的数据生产以及小的体积的优势转变为资本过程中。最近,英国Stanley Royd Hospital的研究人员使用MinION用于监测沙门氏菌(Salmonella enterica)的爆发103。MinION测序仪最令人振奋的应用可能就是2014年的埃博拉病毒爆发104。在位于日内瓦的欧洲移动实验室的主持下,作者对埃博拉病毒的传播以及进化历史进行了深入的研究。
结尾
我们正处在新的NGS技术革命的顶端。NGS现在已经不仅仅只是一个新奇的事物,而已经成为了一个在生物学研究中广泛应用的技术。最新的超高通量测序仪已经将曾经认为不可能的事情成为可能。这包含了首创的精准医疗(medicine initiatives)以及Illumina计划的对循环肿瘤DNA(circulating tumour DNA, ctDNA)进行测序。每个计划都对数万个基因组样本进行测序。所以,快速以及低成本的测序给予了内科医生强大的工具来翻译基因组信息成为有用的临床诊断结果。
这个革命也带来了新的挑战。由于NGS旨在广泛的应用于临床,时间就成为一个NGS首先需要面对的挑战。对于那些严重的神经性疾病或者极为危险的癌症患者而言,数周的WGS分析的等待时间足以使的患者错过最佳的治疗时间。对于急性感染而言,这些事件已经下降到几天。尽管人们已经对时间做出了巨大的改进,但是绝大多数现有的系统都不能完全满足快速模式下的足够产出。
虽然临床诊断面临着数据量不够的问题,NGS其他方面的应用却面临着生产力过剩的境地。目前,已有超过14000个基因组序列上传到US National Center for Biotechnology Information(NCBI)中。2013年,Schatz与Langmead报道了全世界每年可以生产超过15pb的数据量,并且数量与通量依然在继续增加107。数据量的富余对分析以及其下游提出了严峻的挑战,这需要革命性的存储与生信解决方案108。将海量的数据量翻译成有生物学与遗传学内涵的结果同样也是一个挑战87,109,110。在临床诊断方面,通过NGS分析的数据产生的假阳性或者假阴性同样也是需要慎重考虑的问题111,112。
最近,Illumina由于NGS与其周边产品获得了巨大的成功。其它生产商也在快速革新自身的产品113。Illumina的市场仍然在增长,以至于优势巨大。BGISEQ-500以及Helicos technology的GenoCare114在亚洲也有所斩获。ONT PromethION115与Illumina HiSeq X系列则向着成本与产量的极限大步迈进。随着人们对临床诊断测序兴趣的增加,已有的NGS供应商正在提供各种快速的解决方案,如Ion Torrent S5以及Illumina的MiniSeq,还有新加入者Qiagen的GeneReader也来参与竞争。
今后的几年里,更多的玩家也会带着心得解决方案进入这个市场。GenapSys (Sigma-Aldrich)的electronic ‘lunchbox’-sized sequencer116; Genia (Roche)的新的nanopore测序方案117; 以及单通道CMOS技术118,都号称能够在临床应用上节约足够的时间。这些已有的和新的搅局者都有着科技革命的潜质,包括直接对RNA或者蛋白进行测序等,这些最近和未来的进步使得今天成为NGS发展的黄金时期。(来源:生物探索)
Coming of age: ten years of next-generation sequencing technologies
Abstract Since the completion of the human genome project in 2003, extraordinary progress has been made in genome sequencing technologies, which has led to a decreased cost per megabase and an increase in the number and diversity of sequenced genomes. An astonishing complexity of genome architecture has been revealed, bringing these sequencing technologies to even greater advancements. Some approaches maximize the number of bases sequenced in the least amount of time, generating a wealth of data that can be used to understand increasingly complex phenotypes. Alternatively, other approaches now aim to sequence longer contiguous pieces of DNA, which are essential for resolving structurally complex regions. These and other strategies are providing researchers and clinicians a variety of tools to probe genomes in greater depth, leading to an enhanced understanding of how genome sequence variants underlie phenotype and disease.
原文链接:http://www.nature.com/nrg/journal/v17/n6/abs/nrg.2016.49.html
